 Open Access Article
Open Access ArticleFreeze-drying yields stable and pure amorphous calcium carbonate (ACC)†
Johannes
Ihli
*,
Alexander N.
Kulak
and
Fiona C.
Meldrum
*
School of Chemistry, University of Leeds, Leeds, LS2 9JT, UK. E-mail: F.Meldrum@leeds.ac.uk; Fax: +44 (0)113 343 6565; Tel: +44 (0)113 343 6414
First published on 26th February 2013
Abstract
A simple synthetic method is presented for the precipitation of high purity, dry amorphous calcium carbonate (ACC) based on freeze-drying saturated, counter ion free CaCO3 solutions, where the ACC produced shows an extended atmospheric stability. Translation of the methodology to amorphous calcium phosphate demonstrates the generality of the approach.
Since the discovery that the calcite spicules of sea urchin larvae form via an amorphous calcium carbonate (ACC) precursor phase rather than by ion-by-ion growth,1 there has been an explosion of interest in amorphous precursor phases such as ACC and amorphous calcium phosphate (ACP). These are now recognised to be of major importance to the formation and properties of biominerals.2–4 Indeed, the advantages of a crystallisation pathway utilizing amorphous intermediates is showcased by the formation of the remarkable collagen/hydroxyapatite composite that is bone,5 and by the controlled, yet rapid crystallization of mesocrystalline spicules in sea urchins.6 Here, the spicule appears to retain a “memory” of the original ACC particles from which it formed, contributing to its superior mechanical properties.7
There is therefore enormous potential for amorphous precursor phases to be exploited in materials synthesis, with their promise of superior control over nucleation and growth and access to rapid growth rates and “non-crystalline” morphologies.8,9 In order for us to fully profit from these it is essential that their structures and crystallization mechanisms are fully characterised, and we must learn how to control their crystallization. However, we are currently hindered from reaching this goal by problems in synthesising well-defined “reference” ACC samples.
Many methods have been reported leading to the precipitation of ACC,10–12 but these are often poorly reproducible due to the subsequent steps involved in isolating the ACC. Indeed, ACC actually appears to represent a family of phases whose structure and composition is dependent on the particular synthesis method and solution conditions (e.g. temperature, pH).13,14 Treatments following precipitation such as drying or washing with agents such as ethanol15 can also make significant changes to the ACC and its crystallisation behaviour. Consequently, synthetic ACC can vary considerably in terms of stability,16,17 co-precipitated ions, and the amount of structural and surface water,11 which makes characterization of the mechanism of its crystallization difficult. Indeed, synthetic ACC with long-term stability is only typically reported on precipitation from ethanol,15 or in the presence of Mg2+.18
In this work we address this problem and describe a highly reliable synthesis of ACC – based on freeze-drying – which overcomes many of these problems. The ACC is generated in the absence of any counter-ions, contains little surface-bound water and shows an extended atmospheric stability. The generality of this approach for the synthesis of amorphous phases is then demonstrated by extension to ACP.
A schematic diagram of the production of ACC by freeze-drying is shown in Fig. 1. In this technique, it is suggested that ACC forms during the freezing process itself, and that subsequent application of a vacuum leads to sublimation of the ice, generating dry ACC. On placing a solution of calcium carbonate into liquid nitrogen, rapid freezing occurs. During this process the reacting ions, calcium and “carbonate”, are pushed away from the ice front into smaller and smaller liquid reservoirs as freezing progresses, creating highly supersaturated environments in which ACC ultimately precipitates. The ACC is stabilised against crystallisation due to the low temperature and complete solidification of the solvent, and subsequent sublimation of the frozen solvent then releases the as-formed ACC.19,20 That ACC is formed during the freezing process itself is supported by the fact that if slow freezing rates are employed, calcite is obtained rather than ACC.
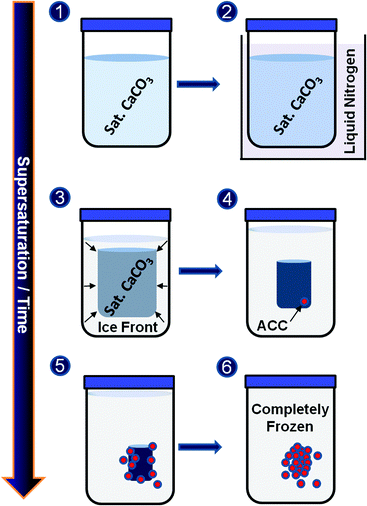 | ||
| Fig. 1 A schematic diagram of the production of ACC by freeze drying. (1) The initial saturated CaCO3 solution is (2) plunged into liquid nitrogen such that it begins to freeze, (3) creating highly supersaturated liquid reservoirs, (4–5) leading to ACC precipitation and (6) followed by final solvent solidification and ACC stabilization. | ||
Synthesis of ACC by freeze-drying necessitates the initial production of “counter-ion free”, saturated CaCO3 solution. “Pure” CaCO3 was formed by repeated dissolution and re-precipitation of CaCO3 which had been obtained by mixing 1 M CaCl2 with 1 M Na2CO3. Saturated solutions of CaCO3 were then prepared by adding 2.5 g of the pure CaCO3 to 500 ml Milli-Q water. These solutions were stored 24 hours at room temperature before centrifuging to remove any remaining solid. Finally, this saturated solution was cooled to remove possible ghost nuclei, before filtering through a 0.2 μm membrane. Freezing of saturated solutions (1–400 ml) was achieved by slow immersion into a liquid nitrogen bath, at a rate of ∼0.5 cm min−1 to give visual solidification in ≤15 min, followed by a 10 min annealing period to consolidate the ice structure. Subsequent sublimation of the excess solvent under vacuum delivers the pure, dry ACC. Immersion rates resulting in solidification in ≥30 min yielded calcite.
The ACC prepared by freeze-drying was characterized using a range of techniques, and was compared with ACC produced by direct mixing of 1 M CaCl2–Na2CO3, followed by filtration, washing with ethanol and air-drying (termed “direct precipitation”). Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) (Fig. 2) confirmed the formation of spherical particles ∼100 nm in diameter, this was confirmed using dynamic light scattering (Fig. S1, ESI†). Powder XRD (Fig. S2A, ESI†) and electron diffraction confirmed that the CaCO3 precipitated by freeze-drying was amorphous, as shown by the presence of a broad amorphous background and the complete absence of any sharp peaks. Energy-dispersive X-ray spectroscopy confirmed the ACC to be virtually counter ion-free (particularly Cl− and Na+) (Fig. S2B, ESI†).
 | ||
| Fig. 2 (A) TEM, and (B) SEM images of ACC produced via freeze-drying. | ||
IR spectroscopy also yielded spectra characteristic of ACC (Fig. 3A) with broad peaks centred around 1473/1406 cm−1 (ν3), 1130 cm−1 (ν1) and 868 cm−1 (ν2), in addition to peaks due to the vibration of water molecules at 1651 cm−1. The notable absence of the ν4 peak around 700 cm−1 further demonstrated the formation of ACC.21 Raman spectroscopy confirmed this analysis and showed a very broad peak centred at 1088 cm−1 (ν4) corresponding to the internal CO32− symmetric stretch (Fig. 3B). Comparison of the Raman spectra of the ACC samples prepared by freeze-drying and direct precipitation showed an increase in peak broadening of the ν4 band of >20% at full width half maximum (FWHM) for the freeze-dried ACC, which is indicative of a higher degree of disorder.22 This conclusion is also supported by the IR spectra, where the freeze-dried ACC exhibits a much stronger symmetric-stretch ν1 peak, observed in non-centrosymmetric structures.3
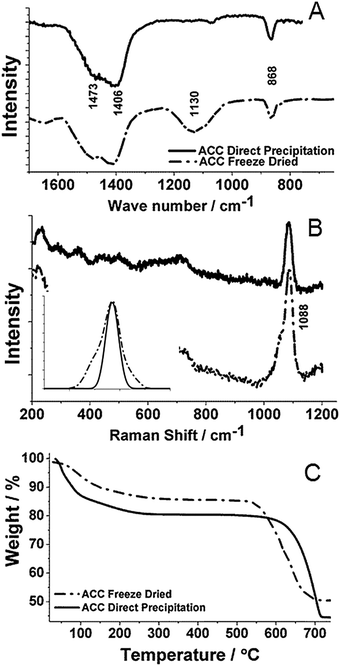 | ||
| Fig. 3 (A) IR spectra, (B) Raman spectra and (C) TGA of freeze-dried ACC and ACC obtained by mixing 1 M CaCl2–Na2CO3. The inset in (B) shows a comparison of the peak widths of both samples. | ||
Thermogravimetric analysis (TGA) was used to investigate the composition of the ACC samples (Fig. 3C). The heating profile of freeze-dried ACC which had been stored in laboratory atmosphere for 2 weeks demonstrated a loss of 10 wt% below 200 °C, corresponding to adsorbed surface water and structural water. This was followed by a continuous weight loss due to CO2 release, before decomposition to CaO occurred above 550 °C. Analysis of the data suggested a sample composition of ∼1CaCO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.7H2O. This can be compared with the ACC produced by direct precipitation which exhibits a loss of ∼20 wt% below 200 °C and a composition of ∼1CaCO3:1H2O. This is typical of ACC synthesised in aqueous solution.10,14
:0.7H2O. This can be compared with the ACC produced by direct precipitation which exhibits a loss of ∼20 wt% below 200 °C and a composition of ∼1CaCO3:1H2O. This is typical of ACC synthesised in aqueous solution.10,14
The freeze-dried ACC also showed remarkable stability such that it was stable against crystallisation under normal atmospheric conditions for up to six weeks, as demonstrated by Raman spectroscopy (Fig. S3, ESI†). We stress that this is achieved without quenching the transformation using ethanol (the standard method used) which partially exchanges with surface-bound water. By contrast, ACC prepared by combination of 1 M CaCl2–Na2CO3 and washed with isopropanol crystallised within 24–48 hours.23
Freeze-drying therefore appears to offer a highly attractive method for producing dry, amorphous phases. To confirm the generality of this approach, the method was also successfully extended to the synthesis of amorphous calcium phosphate (ACP). A counter-ion free saturated solution of calcium phosphate was freeze-dried and the product material characterized. SEM analysis revealed a particle size of ∼50 nm (Fig. 4) and ACP formation was confirmed using Raman and IR-spectroscopy and XRD (Fig. S4, ESI†).24,25 TGA revealed a Ca/P ratio of ∼1.70 indicating a composition of Ca1.7(PO4)1(OH)0.4·3H2O which is similar to hydroxyapatite (Fig. S5, ESI†).
 | ||
| Fig. 4 SEM image and corresponding EDX (Energy Dispersive X-ray) spectrum (inset) of amorphous calcium phosphate (ACP) obtained via freeze-drying. | ||
In summary, we have developed a procedure, based on freeze-drying a counter-ion free solution of a mineral, which yields pure amorphous mineral phases with high reproducibility. Of particular significance, these amorphous solids exhibit low levels of physisorbed and structural water, which in the case of ACC imparts extended atmospheric stability. This indicates that the rapid atmospheric crystallization observed for ACC precipitated from aqueous solution derives from excessive surface water, and that routine variations in the amounts of bound water and ethanol (from washing) may be the origin of many of the problems in obtaining ACC with reproducible properties. This freeze-dried ACC will now be used as the basis for systematic studies of the structure and crystallization mechanisms of ACC, which will ultimately enable us to profit from this phase synthetically.
We thank the EPSRC for financial support via grant numbers EP/H005374/1 (JI and FCM) and EP/G00868X/1 (AK).
Notes and references
- E. Beniash, J. Aizenberg, L. Addadi and S. Weiner, Proc. R. Soc. London, Ser. B, 1997, 264, 461–465 CrossRef CAS
.
- E. Beniash, R. A. Metzler, R. S. K. Lam and P. Gilbert, J. Struct. Biol., 2009, 166, 133–143 CrossRef CAS
- L. Addadi, S. Raz and S. Weiner, Adv. Mater., 2003, 15, 959 CrossRef CAS
-
S. Weiner and L. Addadi, in Annu. Rev. Mater. Res., ed. D. R. Clarke and P. Fratzl, Annual Reviews, Palo Alto, 2011, pp. 21–40 Search PubMed
- E. Beniash, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2011, 3, 47–69 CrossRef CAS
- Y. Politi, T. Arad, E. Klein, S. Weiner and L. Addadi, Science, 2004, 306, 1161–1164 CrossRef CAS
- J. Seto, Y. Ma, S. A. Davis, F. Meldrum, A. Gourrier, Y.-Y. Kim, U. Schilde, M. Sztucki, M. Burghammer, S. Maltsev, C. Jäger and H. Cölfen, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 3699–3704 CrossRef CAS
- Y. Y. Kim, N. B. J. Hetherington, E. H. Noel, R. Kroger, J. M. Charnock, H. K. Christenson and F. C. Meldrum, Angew. Chem., Int. Ed., 2012, 50, 12572–12577 CrossRef
- A. S. Finnemore, M. R. J. Scherer, R. Langford, S. Mahajan, S. Ludwigs, F. C. Meldrum and U. Steiner, Adv. Mater., 2009, 21, 3928–3932 CrossRef CAS
- J. D. Rodriguez-Blanco, S. Shaw and L. G. Benning, Mineral. Mag., 2008, 72, 283–286 CrossRef CAS
- M. Huber, W. J. Stark, S. Loher, M. Maciejewski, F. Krumeich and A. Baiker, Chem. Commun., 2005, 648–650 RSC
- K. Lee, W. Wagermaier, A. Masic, K. P. Kommareddy, M. Bennet, I. Manjubala, S.-W. Lee, S. B. Park, H. Cölfen and P. Fratzl, Nat. Commun., 2012, 3, 725 CrossRef
- N. Koga, Y. Nakagoe and H. Tanaka, Thermochim. Acta, 1998, 318, 239–244 CrossRef CAS
- J. Ihli, Y.-Y. Kim, E. H. Noel and F. C. Meldrum, Adv. Funct. Mater., 2013 DOI:10.1002/adfm.201201805
- K. K. Sand, M. Yang, E. Makovicky, D. J. Cooke, T. Hassenkam, K. Bechgaard and S. L. S. Stipp, Langmuir, 2010, 26, 15239–15247 CrossRef CAS
- H. S. Lee, T. H. Ha and K. Kim, Mater. Chem. Phys., 2005, 93, 376–382 CrossRef CAS
- M. Kellermeier, E. Melero-García, F. Glaab, R. Klein, M. Drechsler, R. Rachel, J. M. García-Ruiz and W. Kunz, J. Am. Chem. Soc., 2010, 132, 17859–17866 CrossRef CAS
- J. Jiang, M. R. Gao, Y. H. Qiu and S. H. Yu, Nanoscale, 2010, 2, 2358–2361 RSC
- E. L. Himri, M. E. L. Himri and A. Núñez, J. Mater. Environ. Sci., 2011, 2, 6 Search PubMed
- D. Vie, E. Martínez, F. Sapiña, J.-V. Folgado, A. Beltrán, R. X. Valenzuela and V. Cortés-Corberán, Chem. Mater., 2004, 16, 1697–1703 CrossRef CAS
- N. V. Vagenas, A. Gatsouli and C. G. Kontoyannis, Talanta, 2003, 59, 831–836 CrossRef CAS
- E. H. Noel, Y.-Y. Kim, J. M. Charnock and F. C. Meldrum, CrystEngComm, 2013, 15, 697–705 RSC
- J. D. Rodriguez-Blanco, S. Shaw and L. G. Benning, Nanoscale, 2011, 3, 265–271 RSC
- S. Gadaleta, E. Paschalis, F. Betts, R. Mendelsohn and A. Boskey, Calcif. Tissue Int., 1996, 58, 9–16 CrossRef CAS
- T. Tsuji, K. Onuma, A. Yamamoto, M. Iijima and K. Shiba, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16866–16870 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, additional IR, Raman and powder XRD spectra. See DOI: 10.1039/c3cc40807h |
| This journal is © The Royal Society of Chemistry 2013 |