 Open Access Article
Open Access ArticleTranslating the concept of peptidelabeling with 5-deoxy-5-[18F]fluororibose into preclinical practice: 18F-labeling of Siglec-9 peptide for PET imaging of inflammation†
Xiang-Guo
Li
*ab,
Anu
Autio
b,
Helena
Ahtinen
b,
Kerttuli
Helariutta
c,
Heidi
Liljenbäck
bd,
Sirpa
Jalkanen
e,
Anne
Roivainen
*bd and
Anu J.
Airaksinen
*c
aDepartment of Pharmacology, Drug Development and Therapeutics, University of Turku, FI-20014 Turku, Finland. E-mail: xiali@utu.fi
bTurku PET Centre, University of Turku and Turku University Hospital, FI-20521 Turku, Finland. E-mail: anne.roivainen@utu.fi
cLaboratory of Radiochemistry, Department of Chemistry, University of Helsinki, FI-00014 Helsinki, Finland. E-mail: anu.airaksinen@helsinki.fi
dTurku Center for Disease Modeling, University of Turku, FI-20014 Turku, Finland
eMediCity Research Laboratory and Department of Medical Microbiology and Immunology, University of Turku, FI-20014 Turku, Finland
First published on 15th March 2013
Abstract
Peptide glycosylation with 5-deoxy-5-[18F]fluororibose was translated into preclinical settings. The novel 18F-labeled Siglec-9 peptide was produced using an automated synthesis procedure. The 18F-labeled Siglec-9 peptide showed favorable binding in the animal model of inflammation in vivo.
Positron emission tomography (PET) is a sensitive and non-invasive in vivo molecular imaging modality. Fluorine-18 (18F) has favorable physical properties to generate high-resolution PET images and consequently 18F-labeled compounds are the most used tracers in clinical PET. Among the short-lived radionuclides, the physical half-life of 18F (T1/2 = 109.7 minutes) allows multi-step synthesis of tracers for the same-day use in hospitals.1 Versatile fluorinating agents have been developed for the synthesis of fluorine containing pharmaceuticals.1 However, only a small proportion of the existing fluorination methods have been (or can be) translated into radiosynthesis of 18F-labeled compounds for preclinical and clinical applications.1a This is especially true in the case of 18F-labeled biomolecules, e.g.peptides, proteins and antibodies. Although many strategies for 18F-labeling of peptides and proteins have shown some promise during the last two decades, only a few have proved amenable for routine large scale production for clinical use. Oxime formation between an aldehyde (or a ketone) and an aminooxy-functionalized compound is a classical and favorable reaction in bioconjugation due to the high chemoselectivity. Indeed, oxime formation has been successfully applied for the production of [18F]fluciclatide (GE Healthcare) starting from 4-[18F]fluorobenzaldehyde ([18F]FBA). [18F]Fluciclatide is one of the few 18F-labeled peptidetracers currently in clinical use.1b Among the 18F-labeled aldehydes, [18F]FBA is probably the most frequently used prosthetic group for peptidelabeling. However, [18F]FBA is volatile requiring additional measures to ensure radiosafety and to reduce radioactive emission during the production. The attachment of [18F]FBA may cause increased lipophilicity, resulting in altered pharmacokinetics in vivo.1b In addition to [18F]FBA, aldehydes1–5 have recently been developed for peptidelabeling (Fig. 1, [a]).1b,2
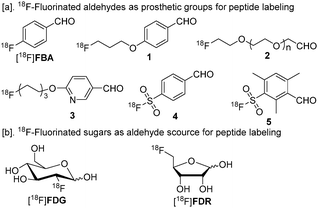 | ||
| Fig. 1 18F-Labeled prosthetic groups for oxime formation.18F-Fluorinated sugars are presented in their cyclic forms.1b,2 | ||
It has been well documented that glycosylation may confer favorable characteristics to peptidetracers for in vivo imaging. The incorporation of a galactose fragment into the RGD peptide has generated a very valuable peptidetracer, [18F]galacto-RGD. So far, most of the published clinical PET imaging data concerning 18F-labeled peptides has actually been generated using this tracer. However, the synthesis procedure of [18F]galacto-RGD is rather complex and is a major challenge for its widespread use.3 As 2-deoxy-2-[18F]fluoroglucose ([18F]FDG, Fig. 1, [b]) is available in virtually every PET radiochemistry lab, it is an excellent concept to use [18F]FDG for peptide and proteinlabeling.4 [18F]FDG is an aldose and has an open chain aldehyde form in solution.1b However, the conjugation between [18F]FDG and aminooxy-functionalized peptides generally needs elevated temperature and low pH and forms a complex product profile.1b,4b Very recently, it has been discovered that 5-deoxy-5-[18F]fluororibose ([18F]FDR, Fig. 1, [b]) conjugates to peptides much more efficiently than [18F]FDG does.5 Although we were able to produce [18F]FDR in large scale in an automated procedure, we were not able to perform the conjugation step efficiently enough for preclinical PET studies. Furthermore, the quality of radiolabeled peptides was not acceptable (e.g. low chemical purity) for in vivo animal studies. Thus, peptideglycosylation with [18F]FDR remained at a conceptual stage with respect to preclinical and clinical utilities. In this communication, we report the translation of [18F]FDR-based bench chemistry into practical preclinical use, and exemplify this utility on PET imaging of vascular adhesion protein 1 (VAP-1), which is a promising new target protein for PET imaging of inflammation.
In the previous work, [18F]FDR was prepared in high radiochemical purity (>98%) from precursor 6 (Scheme 1, [a]), and the synthesis was scaled up to 4 GBq of radioactivity. However, chemical purity of the synthesized [18F]FDR was not optimal and the final product contained ribose in a mM-range, originating from precursor 6. Subsequently, only a small proportion of the obtained [18F]FDR (8–20 MBq) was conjugated to model peptides. In the presence of an appropriate amount of peptides (e.g. at 23 mM of concentration), the conjugation reached completion in 10 minutes in sodium acetate buffer (pH 4.6) at rt.5 Scaling up the conjugation reaction was hampered due to the high ribose content of the produced [18F]FDR. When starting with 100 MBq-4 GBq of [18F]FDR, only 5% conversion yields were achieved in an hour, even at high concentrations of peptide (e.g. 40 mM). Since only nano- or picomoles of non-carrier added and high specific radioactivity [18F]FDR was used, the ribose was present in the conjugation mixture in a large excess. Its presence as a competing aldehyde source resulted in the inefficient conjugation of [18F]FDR (Scheme 1, [b]). This prompted us to derive a strategy to produce [18F]FDR free of ribose ([18F]FDR 8, Scheme 1, [a]). After the fluorination of precursor 6 with K18F-K222, intermediate 7 was isolated using a preparative HPLC column. The method gave an excellent separation between the starting material 6 (retention time = 10.6 min) and intermediate 7 (retention time = 4.9 min) in a short HPLC protocol. Thus, the synthesis procedure was not significantly prolonged. The HPLC analysis of the isolated 7 proved the success of the intermediate purification step (ESI†). After the acid-catalyzed hydrolysis of 7, [18F]FDR 8 was obtained in high radiochemical purity (>98%). Subsequently, the pH of [18F]FDR 8 solution was adjusted to 4.6 at which the efficiency of peptide conjugation was optimal. The total preparation time of the high purity [18F]FDR 8 was typically 85 min starting from end-of-bombardment (EOB), including a concentration step of the [18F]FDR solution before the conjugation reaction. In addition to 6, 1,2,3-tri-O-acetyl-5-O-tosyl ribose (AcRib, ESI†) was tested as a precursor to produce [18F]FDR 8. Unfortunately, only residual amount of [18F]FDR 8 was obtained from AcRib. In this respect, compound 6 was a very applicable precursor in radiosynthesis (n = 40). The next step was to prepare VAP-1 targeting [18F]FDR-Siglec-9 peptide for in vivo imaging of inflammation in rats (Scheme 1, [a] and [c]). VAP-1 is a 90 kDa glycoprotein and is a unique target in inflammation.6 Inflammation is highly relevant in major diseases including cancer, diabetes, obesity and Alzheimer's disease.6b In endothelial cells, VAP-1 stays in the intracellular storage granules under normal physiological conditions. Upon inflammation, VAP-1 relocates rapidly to the endothelial cell surface. This makes VAP-1 an ideal target for diagnostic imaging of inflammation. Recently, it has been discovered that sialic acid-binding Ig-like lectin 9 (Siglec-9) is a leukocyte ligand of VAP-1 and 68Ga-labeled Siglec-9 motif peptide can be used as a PET tracer for in vivo imaging of inflammation and cancer.7 Accordingly, peptide9 (ESI†) was designed in order to enable conjugation to [18F]FDR 8 in this work.
![Synthesis and preclinical utilization of [18F]FDR-Siglec-9.](/image/article/2013/CC/c3cc40738a/c3cc40738a-s1.gif) | ||
| Scheme 1 Synthesis and preclinical utilization of [18F]FDR-Siglec-9. | ||
In general, oxime formation is not the most efficient chemical reaction and the substrates need to be used at relatively high concentrations in order to reduce the reaction time. Specific radioactivity of the cyclotron produced 18F is high and the amount of the synthesized [18F]FDR 8 is in a nano- to picomolar level. Following the reported conjugation procedure,5 the initial tests for the synthesis of [18F]FDR-Siglec-9 peptide were carried out in sodium acetate buffer (pH 4.6, 90 mM) at rt. However, the concentration of peptide9 used (2.1 kDa) had to be at over 15 mM (32.2 mg ml−1), which was absolutely not applicable. In a previous study, aniline was used to catalyze the oxime formation between [18F]FBA and leptin.8 Accordingly, we tested aniline-catalysis in the conjugation reactions between 5-deoxy-5-fluororibose ([19F]FDR) and 9 (Table 1). Being the “cold” counterpart of [18F]FDR 8, [19F]FDR was used to prepare [19F]FDR-Siglec-9, which was needed as a reference in radiosynthesis of [18F]FDR-Siglec-9. In anilinium buffer (0.3 M, pH 4.6), the conjugation efficiency was dramatically enhanced (Table 1, entries 1 and 2). Aniline has been proposed to catalyze oxime formation with a rapid transimination mechanism.1b The input concentration of peptide9 could be as low as 0.3 mM (0.6 mg ml−1) and the conversion was 95% in 10 min at rt. The concentration of the anilinium buffer also had an influence on the conjugation efficiency (entries 2–4). Our results together with the previous work demonstrated that aniline-catalyzed oxime formation is chemoselective in the presence of any amino acid side chain from all the 20 standard amino acids under 18F-labeling conditions.8
| Entry | Concentration of [19F]FDR and 9a (mM) | Buffer (M) | Conversionb (%) |
|---|---|---|---|
| a The two reagents were used in equal amounts. b Conversions were determined using HPLC. | |||
| 1 | 0.3 | Sodium acetate (0.3) | 5 |
| 2 | 0.3 | Anilinium acetate (0.3) | 95 |
| 3 | 0.3 | Anilinium acetate (0.2) | 89 |
| 4 | 0.3 | Anilinium acetate (0.05) | 63 |
| 5 | 0.1 | Anilinium acetate (0.3) | 48 |
A general challenge in developing peptide PET tracers is to isolate the labeled products (e.g. [18F]FDR-Siglec-9) from their unlabeled peptide precursors (e.g.9). The possible residual peptide precursors may compete with the tracer on binding to the target protein or even saturate the target to be imaged. Bearing this in mind, we developed a reproducible HPLC method for efficient separation of [18F]FDR-Siglec-9 from 9 in a preparative scale (ESI†). Finally, [18F]FDR-Siglec-9 was prepared from the conjugation of [18F]FDR 8 to 9 (0.3 mM) in anilinium buffer (pH 4.6) at rt and the conversion was 50–60% in 10 min. The final product was isolated by HPLC with high radiochemical purity (>98%). The amount of the peptide precursor 9 was under the detection limit of the UV-detection. The total synthesis time was typically 120 minutes and the decay-corrected radiochemical yield was 27% starting from 18F-fluoride (EOB). The two HPLC-purifications were well integrated in the protocol and were performed with remote-control. Both radiation exposure and radioactive emission were minimal. The synthesis was scaled up to 1.1 GBq of the final product [18F]FDR-Siglec-9. The specific radioactivity was 36–43 GBq μmol−1 at the end-of-synthesis (EOS) (ESI†). [18F]FDR-Siglec-9 was formulated in phosphate-buffered saline (PBS) for intravenous injection. In PBS, [18F]FDR-Siglec-9 was stable for >4 hours and thus suitable for in vivo imaging studies. The [18F]FDR-Siglec-9 (18.3 ± 5.1 MBq) was intravenously injected into rats having sterile, turpentine oil induced inflammation.7 Dynamic PET imaging lasting for 1 hour was performed by using a High Resolution Research Tomograph (Siemens). The inflammation focus on the right shoulder area of the rats (n = 8) was clearly visualized (Fig. 2). The target-to-muscle ratio was 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The observed liver uptake and rapid excretion through kidneys to the urine were in line with our previous studies with [68Ga]-DOTA-Siglec-9.7 Very importantly, bones were not visualized, which was an indication that there was no in vivo defluorination of the 18F-label on the prosthetic group [18F]FDR 8.
:1. The observed liver uptake and rapid excretion through kidneys to the urine were in line with our previous studies with [68Ga]-DOTA-Siglec-9.7 Very importantly, bones were not visualized, which was an indication that there was no in vivo defluorination of the 18F-label on the prosthetic group [18F]FDR 8.
![Representative sagittal (left), transaxial (middle) and coronal (right) multiplane PET images of [18F]FDR-Siglec-9 biodistribution in a rat. The images are summation from 10–60 min post-injection.](/image/article/2013/CC/c3cc40738a/c3cc40738a-f2.gif) | ||
| Fig. 2 Representative sagittal (left), transaxial (middle) and coronal (right) multiplane PET images of [18F]FDR-Siglec-9 biodistribution in a rat. The images are summation from 10–60 min post-injection. | ||
In conclusion, we have successfully translated [18F]FDR-based oxime formation into the production of [18F]FDR-Siglec-9 peptide for preclinical use. [18F]FDR-Siglec-9 has been produced in high radiochemical quality and sufficient specific radioactivity facilitating in vivo PET imaging of experimental inflammation. Further development of the tracer for clinical use is warranted. [18F]FDR-conjugation is independent of peptide sequence and the whole synthesis procedure can be automated. Thus, [18F]FDR-based glycosylation should be generally applicable in the development of peptide PET tracers.
We thank the Academy of Finland (no. 133127, 136805, 119048 and 258814) for financial support. The study was conducted within the Finnish Centre of Excellence in Molecular Imaging in Cardiovascular and Metabolic Research supported by the Academy of Finland, the University of Turku, the Turku University Hospital and the Åbo Akademi University.
Notes and references
- (a) M. Tredwell and V. Gouverneur, Angew. Chem., Int. Ed., 2012, 51, 11426 CrossRef CAS; (b) X.-G. Li, M. Haaparanta and O. Solin, J. Fluorine Chem., 2012, 143, 49 CrossRef CAS , and references therein; (c) X.-G. Li, J. Domarkas and D. O'Hagan, Chem. Commun., 2010, 46, 7819 RSC.
- J. A. H. Inkster, K. Liu, S. Ait-Mohand, P. Schaffer, B. Gurin, T. J. Ruth and T. Storr, Chem.–Eur. J., 2012, 18, 11079 CrossRef CAS.
- A. J. Beer and M. Schwaiger, J. Nucl. Med., 2011, 52, 335 CrossRef CAS.
- (a) O. Boutureira, G. J. L. Bernardes, F. D'Hooge and B. G. Davis, Chem. Commun., 2011, 47, 10010 RSC; (b) C. Hultsch, M. Schottelius, J. Auernheimer, A. Alke and H.-J. Wester, Eur. J. Nucl. Med. Mol. Imaging, 2009, 36, 1469 CrossRef CAS.
- X.-G. Li, S. Dall'Angelo, L. F. Schweiger, M. Zanda and D. O'Hagan, Chem. Commun., 2012, 48, 5247 RSC.
- (a) M. Salmi and S. Jalkanen, Science, 1992, 257, 1407 CAS; (b) A. Roivainen, S. Jalkanen and C. Nanni, Eur. J. Nucl. Med. Mol. Imaging, 2012, 39(suppl. 1), S68 CrossRef; (c) Http://www.biotie.com .
- K. Aalto, A. Autio, E. A. Kiss, K. Elima, Y. Nymalm, T. Z. Veres, F. Marttila-Ichihara, H. Elovaara, T. Saanijoki, P. R. Crocker, M. Maksimow, E. Bligt, T. A. Salminen, M. Salmi, A. Roivainen and S. Jalkanen, Blood, 2011, 118, 3725 CrossRef CAS.
- R. R. Flavell, P. Kothari, M. Bar-Dagan, M. Synan, S. Vallabhajosula, J. M. Friedman, T. W. Muir and G. Ceccarini, J. Am. Chem. Soc., 2008, 130, 9106 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Radiosynthesis of ribose-free [18F]FDR 8 and [18F]FDR-Siglec-9 and PET imaging in rats. See DOI: 10.1039/c3cc40738a |
| This journal is © The Royal Society of Chemistry 2013 |