 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Conformationally preorganised hosts for anions using norbornane and fused [n]polynorbornane frameworks
Adam J.
Lowe†
,
Benjamin M.
Long
and
Frederick M.
Pfeffer
*
Research Centre for Chemistry and Biotechnology, School of Life and Environmental Sciences, Deakin University, Waurn Ponds, VIC 3217, Australia. E-mail: fred.pfeffer@deakin.edu.au; Fax: +61 3 5227 1040; Tel: +61 3 5227 1439
First published on 13th February 2013
Abstract
Norbornane and fused [n]polynorbornane frameworks are readily synthesised, can be tailored to a variety of predictable geometries and can be functionalised regiospecifically. As such, these highly preorganised scaffolds offer the supramolecular chemist an excellent starting point when designing hosts for specific guests. This feature article will highlight the evolution of our research from relatively simple norbornane based anion receptors to more sophisticated tetrathioureido functionalised fused [n]polynorbornane hosts.
 Adam J. Lowe | Adam Lowe completed his PhD under the supervision of Dr Pfeffer in 2009 on the development of norbornenes and [n]polynorbornanes as molecular scaffolds for anion recognition. He is currently senior scientist in the digital biology center of Bio-Rad Laboratories Pty., Ltd. California where he is developing new amphiphiles for use in digital droplet technology. |
 Benjamin M. Long | Benjamin Long received his BSc (Hons) at Deakin University, Geelong, Australia and is currently completing his PhD under the guidance of Dr Frederick Pfeffer. His research focusses on the use of fused norbornane frameworks for anion recognition as well as the functionalisation of these scaffolds as peptidomimetics. |
 Frederick M. Pfeffer | Fred Pfeffer completed his PhD in 2001 on the synthesis of peptide functionalised molecular frameworks before moving to Trinity College Dublin for a teaching post then postdoctoral fellowship with Thorfinnur Gunnlaugsson and Paul Kruger on the development of naphthalimide based anion sensors. He returned to Australia in 2004 to take up a lecturing position at Deakin University where he is now senior lecturer. His interests include the development of new antidiabetic and antimicrobial agents as well as supramolecular anion recognition chemistry; in particular the development of conformationally preorganised norbornane based hosts. |
Introduction
In the field of supramolecular chemistry the topic of anion recognition and sensing has become an intense pursuit for a growing number of research groups worldwide.1 Indeed, as a result of this effort, many excellent examples of hosts for anionic species have been successfully developed.2In many instances new hosts are synthesised and are subsequently evaluated against a broad set of guests to see which of these ‘fits’ best. One of the principal objectives for undertaking the research featured herein was not to simply unveil a new host in this way but to develop a series of related hosts such that the supramolecular chemist, when faced with a specific guest, can employ a host of appropriate dimensions to complement that guest. An approach that employs a framework comprised of n individual norbornane units (1, Fig. 1) fused together to form a [n]polynorbornane framework such as 2 is one that can provide hosts that contain a preorganised cleft of predictable dimensions.
![Norbornane 1 and fused [3]polynorbornane framework 2.](/image/article/2013/CC/c3cc40702k/c3cc40702k-f1.gif) | ||
| Fig. 1 Norbornane 1 and fused [3]polynorbornane framework 2. | ||
This feature article will briefly outline the role of preorganisation in supramolecular chemistry then highlight the recent use of norbornane and in particular, topologically predefined, fused [n]polynorbornanes in the recognition of anionic species.
Preorganisation, induced fit, and complementarity
It was Emil Fischer who, in 1894, noted that “only in the case of similar geometrical structure can the molecules approach each other as to initiate a chemical action…together like a lock and key”.3 This pioneering theory of enzyme:substrate binding was modified by Koshland who introduced a flexible hand in glove description for the topological adjustment—induced fit—that enzyme:substrate complexes undergo in order to achieve the optimum alignment of binding groups.4In the realm of supramolecular chemistry it was Cram who used preorganised spherands and flexible podands to clearly demonstrate that “preorganisation is a central determinant of binding power” and also that the alignment of contact sites between the host and guest—complementarity—is crucial for specific recognition.5 Thus the goal of strong and selective anion recognition by charge neutral hosts can only occur if the host has an array of hydrogen bond donors suitably arranged in a predefined fashion for the guest.
In this context fused [n]polynorbornane frameworks again appear well suited as they can be readily synthesised to specific dimensions and can also be easily functionalised to include a variety of H-bond donors
This article will focus on ‘larger’ scaffolds, nevertheless, researchers will be aware of anion hosts based on ‘smaller’ scaffolds such as pyrrole,6 indole,7 naphthalene,8 naphthalimide,9 and anthracene.10 In addition, suitably functionalised metal templated architectures have also emerged as suitable for anion recognition and the subject has been recently reviewed.11
Norbornanes
The norbornane (bicyclo[2.2.1]heptane) framework 1 requires little introduction. It occurs naturally in terpenoid derivatives such as borneol, camphor and fenchone12 and the cycoladdition methodology developed for its construction won its discoverers, Otto Diels and Kurt Alder, a Nobel prize in 1950.13 This simple scaffold has enjoyed use in a range of fields where conformational preorganisation is paramount including medicinal chemistry,14 peptidomimetics15 and as chiral auxiliaries for asymmetric synthesis.16In the field of supramolecular chemistry tetra-amide norbornenes have been used as photo-switchable ion carriers (Fig. 2).17 Norbornadiene–quadricyclane isomerisation (3a–3b) alters the preorganisation of the four amide groups and has a direct impact on the binding (and in turn transport: organic → aqueous) of a range of cationic guests.17
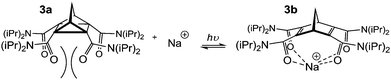 | ||
| Fig. 2 Photoisomerisation of 3a to 3b enables cation recognition and transport from chloroform to water.17 | ||
Norbornanes and anion recognition18,19
To demonstrate that norbornanes/enes could be employed as frameworks for anion recognition, hosts 4–6 were designed (Fig. 3). Each possessed a unique binding cleft flanked by two thiourea arms (throughout this article 2-ureidoethylamido substituents are referred to as arms for convenience).18,19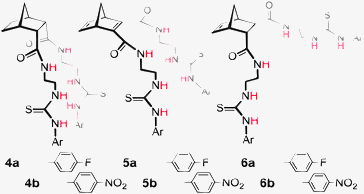 | ||
| Fig. 3 Norbornane and norbornene based hosts 5–7. | ||
Synthesis of the hosts was achieved using Diels–Alder cycloaddition, amide bond formation and thiourea formation. For example, construction of endo/endo host 419 (Scheme 1) required cycloaddition of cyclopentadiene with acetylenedicarboxylic acid to afford norbornadiene diacid 7. Coupling with two equivalents of 2-(tert-butoxycarbonylamino)ethylamine using EDCI gave Boc protected diamide 8. Hydrogenation using Pd(OH)2 afforded norbornane 9 with the desired endo/endo geometry. Deprotection (TFA/CH2Cl2) and reaction with the appropriate isothiocyanate afforded hosts 6a and 6b.
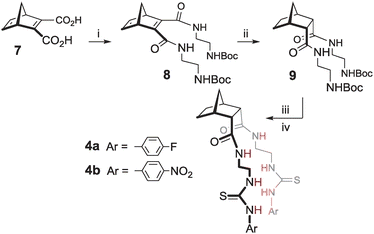 | ||
| Scheme 1 Synthesis of hosts 4. Reagents and conditions: (i) 2-(tert-butoxycarbonylamino)ethylamine, EDC, CHCl3, RT, 17 h, 44% (ii) H2, Pd(OH)2/C, EtOH, RT, 12 h, 99% (iii) 12% TFA/DCM, RT, 3 h, 100% (iv) DIPEA, CHCl3, RT, 18 h, for 4a 4-fluorophenylisothiocyanate, 93%, for 4b 4-nitrophenylisothiocyanate, 88%. | ||
Anion binding studies
Within this series (4–6) it was predicted that compounds 6a and 6b with endo/exo preorganisation would be more suited to tetrahedral anions such as dihydrogenphosphate and these anions would bind in the larger cleft of these hosts in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 host:guest (H:G) arrangement.
:1 host:guest (H:G) arrangement.
To evaluate host:guest interactions, both 1H NMR and UV-Vis titration experiments were performed in DMSO. Selected results from these binding studies are summarised in Table 1.19
:guest (H:G) stoichiometries and calculated association constants (logK) for hosts 5–7a
| 4a | 4b | 5a | 5b | 6a | 6b | ||
|---|---|---|---|---|---|---|---|
|
a logK were determined by 1H NMR titration using WinEQNMR software,20 (error < 14.0%). Titrations were carried out with initial host concentrations, [H]i, of ∼1.2 × 10−2 M. Max Δδ obtained from ArN–H after addition of 5.0 eq. of anion.
|
|||||||
| Cl− | max Δδ (ppm) | 0.51 | 0.53 | 0.48 | 0.84 | 0.44 | 0.53 |
| H:G |
1:2 |
1:2 |
1:2 |
1:2 |
1:2 |
1:2 |
|
| logK1 |
2.8 | 2.5 | 2.6 | 2.9 | 2.3 | 2.6 | |
| logK2 |
1.2 | 1.6 | 1.1 | 1.1 | 1.7 | 1.4 | |
| H2PO4− | max Δδ (ppm) | 1.67 | 1.74 | 1.83 | 1.74 | 1.94 | 1.84 |
| H:G |
1:2 |
1:2 |
1:2 |
1:2 |
1:2 |
1:2 |
|
| logK1 |
3.7 | 3.7 | 3.9 | 2.9 | 3.6 | 3.1 | |
| logK2 |
3.0 | 2.6 | 2.2 | 2.7 | 2.7 | 2.6 | |
| AcO− | max Δδ (ppm) | 3.15 | 3.31 | 2.89 | 3.27 | 3.24 | 3.14 |
| H:G |
1:2 |
1:2 |
1:2 |
1:2 |
1:2 |
1:1 |
|
| logK1 |
3.2 | 3.8 | 4.2 | 3.4 | 3.8 | 3.3 | |
| logK2 |
2.3 | 3.0 | 2.5 | 3.2 | 2.7 | — | |
Electron withdrawing groups and H![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :G stoichiometry
:G stoichiometry
Most significant were the results obtained for acetate (Fig. 4). Five of the six hosts bound this anion in a 1:2 H:G stoichiometry (common for 2-armed thiourea receptors,21 indicating that the urea groups are not acting cooperatively). However, host 6b bound acetate with a 1:1 H:G stoichiometry. The change from Ar–F to the more electron withdrawing Ar–NO2 effected a change in H:G stoichiometry from 1:2 (for 6a ArF) to 1:1 (for 6b ArNO2). Hosts 4b and 5b also contained the NO2 substituent but 1:2 H:G stoichiometry with acetate was identified. This result implied that it was actually a combination of both the endo/exo preorganisation of host 6b and the electron withdrawing nature of the Ar–NO2 that made host 6b unique when binding AcO−.18,19
 | ||
| Fig. 4 Proposed 1:2 H:G binding conformation of host 7a with two equivalents of AcO− and 7b in a 1:1 arrangement with AcO−. | ||
A colour change was noted during the titrations of the hosts containing the nitro group (4b, 5b and 6b) and UV-Vis titrations confirmed the unusual 1:1 H:G stoichiometry of 6b with AcO− despite the initial host concentration being significantly lower (ca. 5.0 × 10−5 M). Association constants calculated from this data were also consistent with those determined from 1H NMR titrations; 4b logK1 = 3.7, logK2 = 3.6; 5b logK1 = 3.7, logK2 = 3.8; and 6b logK1 = 3.9.18,19
To the best of our knowledge this was the first example in which a change in an electron withdrawing group (an Ar–F to a Ar–NO2) could alter the final stoichiometry of the host:guest complex and suggests that H-bonding power can be used to control binding stoichiometry.
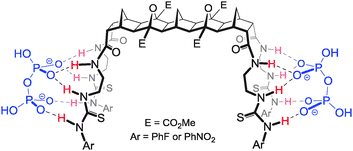
Targeting dihydrogenphosphate and lipid A25
In all cases 1:2 H:G arrangements were noted for the binding of H2PO4− by receptors 4–6 (Fig. 5).18,19 Hosts 4 and 5 bound the guests symmetrically through three H-bonding interactions per arm (two from the thiourea NH's and one from the amide NH), however in the case of the endo/exo host 6 the binding of H2PO4− to the exo arm was by means of three H-bonds, whereas the endo arm bound H2PO4− solely through the thiourea N–H groups.
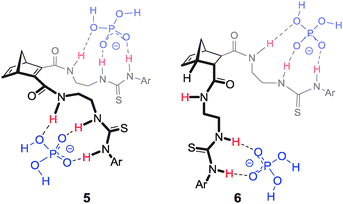 | ||
| Fig. 5 Proposed 1:2 H:G binding conformation of hosts 5 and 6 (also representative of the binding mode of 4) with H2PO4−. | ||
Given that each arm was acting independently in the binding of H2PO4− it was reasoned that this type of host might be capable of binding a diphosphate species such as lipid A.22 (Fig. 6). Many potent antimicrobial agents interact strongly with the anionic lipid A portion of the bacterial outer membrane lipopolysaccharide (LPS). Examples include the naturally occurring polymyxin and defensin families of peptides.23 These compounds are facially amphiphilic; they have cationic groups (ammonium or guanidinium) correctly positioned to interact with the anionic phosphate groups of lipid A and hydrophobic residues to penetrate the hydrophobic layer. In order to mimic these features, ‘lead’ compound 6 was modified to include an octyl ‘tail’ and guanidine groups for anion recognition24 (see 10, Fig. 6). Molecular modelling indicated that the exo/endo arms could easily span lipid A and bind to both phosphate groups.25
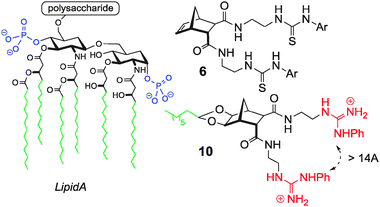 | ||
| Fig. 6 Structure of Lipid A (phosphate groups highlighted in red). Anion host 6 and custom modified host 10 are shown on the right.25 | ||
A fluorescent displacement assay confirmed binding to the LPS target and compound 10 had an IC50 of 9.5 μM (Colistin IC50 = 6.0 μM). Simple disk diffusion studies identified that compound 10 was active (particularly against Pseudomonas aeruginosa ATCC 27853) and haemolytic tests confirmed that 10 did not lyse red blood cells at concentrations up to 125 μM.25
Preorganised frameworks for anion recognition
Excellent progress in the field of anion recognition has been made using conformationally preorganised frameworks.22 Particularly well studied are cholic acid and the calix families.26–36 In the case of the ‘cholapods’ Davis and others27–30 have performed comprehensive studies, varying the number of attachment points and also the number and nature of H-bond donors. Strong binding of chloride was noted for these receptors (e.g.11, Fig. 7)28 with selectivity resulting from a well preorganised binding site. More recent developments include cationic cyclocholamides such as 12 for anion transport.29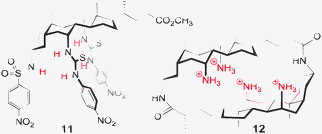 | ||
| Fig. 7 Examples of anion hosts based on cholic acid.28,29 | ||
Many examples also exist of anion hosts based on the calix[n]arenes30–33 and also the calix[n]pyrroles.34–36 The ability to construct related frameworks in varying sizes is an advantage of the calix based hosts and typically [n] = 4 or 6 for these systems (e.g. calix[n]arene, Fig. 8).30 Specific examples, Fig. 8, include hosts for sensing37 (e.g. calix[4]arene 1335) and transport33 (e.g. calix[4]pyrrole 1436).
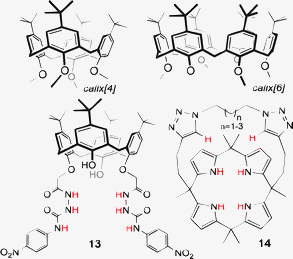 | ||
| Fig. 8 Examples of functionalised calixarene and calixpyrrole frameworks for anion recognition, sensing and transport.35,36 | ||
Other examples of preorganised anion hosts include tripodal benzene receptors38 (e.g.15,39Fig. 9), peptidomimetic trioxazoles40 (e.g.1641) and macrocyclic C–H receptors42 (e.g.1743).
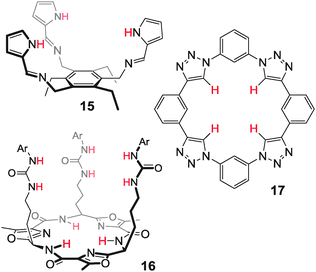 | ||
| Fig. 9 Examples of preorganised tripods and macrocycle.39,41,43 | ||
Fused [n]polynorbornane scaffolds
A collection of researchers including Warrener, Russell, Paddon-Row and Johnston (amongst others) have produced a remarkable arsenal of frameworks of varying, though predictable, dimensions (including tweezers, clips, binanes, molracs, ladderanes, norbornylogs, alicyclophanes, and regioselectively addressable frameworks).44 These versatile scaffolds have been used in a number of settings including (i) single molecule conductivity switching (e.g. tetrasulfide 1845Fig. 10), (ii) DNA bisintercalators to mimic the properties of ditercalinium (e.g. ‘staple-like’ bisacridine 1946) and (iii) molecular capsules (e.g. bisporphyrin tweezers 2047).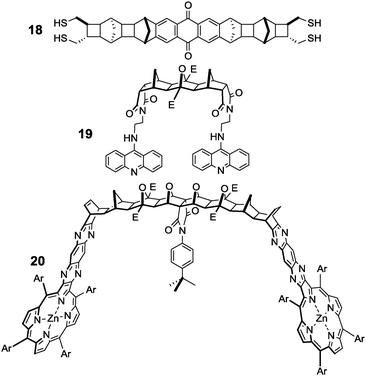 | ||
| Fig. 10 Examples of fused polynorbornane frameworks.45–47 | ||
Cyclic scaffolds that incorporate aryl fused norbornanes have also been pursued by Stoddart48 who employed a molecular LEGO approach to constructing ‘belts’ such as Kohnkene (21,49Fig. 11). More recent examples include the molecular ‘tweezers’ and ‘clips’ produced by Klarner.50 For example bisphosphate 2251 (Fig. 11) binds cationic Lys residues and has been shown to ‘unwind’ amyloidogenic proteins. Fused polynorbornanes have also recently been used by Clever in the synthesis of metal organic cages52 (such as 23,53Fig. 11).
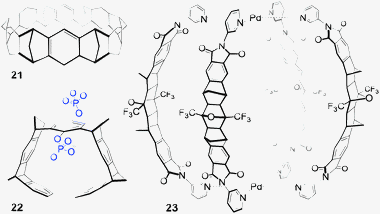 | ||
| Fig. 11 Examples of fused aryl/norbornane frameworks.49,51,53 | ||
Several cycloaddition strategies have been developed to access these fused polynorbornane structures44–55 and inventive descriptions such as LEGO48 are used to describe their construction. Other monikers including ‘molecular glue’ have been used to describe oxadiazole coupling54 and also BLOCK as an acronym for ‘bonzer little organic construction kit’.55 All such terminology hints at the modular nature of the various approaches to the ready assembly of these large molecular architectures. The terminology also clearly conveys the ‘no atoms wasted’ advantage inherent with a cycloaddition approach.56
The ACE reaction
The key cycloaddition to assemble fused [n]polynorbornanes is the (2+3) [π4s + π2s] 1,3 dipolar cycloaddition of a resonance stabilised, electron deficient, carbonyl ylide (such as 24, Scheme 2), generated by electrocyclic ring opening of a cyclobutane epoxide (23), to a norbornene partner (25).57 This reaction of an Alkene with a Cyclobutane Epoxide is termed the ACE reaction. More recently a microwave-assisted version of this cycloaddition has been used to effect the transformation in high yields and reduced reaction times (10–15 minutes).58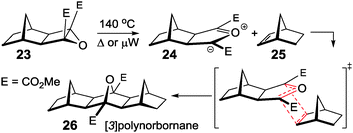 | ||
| Scheme 2 Mechanism of the ACE cycloaddition of an electron deficient cyclobutane epoxide with a norbornene.57,58 | ||
A simple two step methodology has been devised for the construction of the requisite epoxides (Scheme 3). First is the ruthenium catalysed Mitsudo reaction59 of a norbornene with an acetylene dicarboxylate diester (equivalent to a [2+2] cycloadditon) which affords cyclobutene diesters (such as 28). This reaction can be performed in a microwave reactor and near quantitative yields are achieved in under 5 minutes.60 The second step is a modified Weitz–Scheffer epoxidation61 of this electron deficient alkene using tert-butylhydroperoxide (TBHP) with a catalytic amount of potassium tert-butoxide in THF.62 Epoxide 29 and bisepoxide 30 are both routinely used in framework construction and are easily prepared using the protocol of Mitsudo reaction followed by epoxidation.
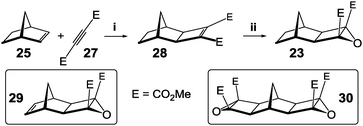 | ||
| Scheme 3 Two step protocol for the synthesis of cyclobutane epoxides. Reagents and conditions: (i) RuH2(CO)(PPh3)3, THF, 80 °C (ii) TBHP, KOtBu, THF, 0 °C. | ||
Fused polynorbornanes are slightly curved in nature (as shown in Fig. 12),63 but linear variants can be accessed through the use of the dihydrofulvalene ‘pincer’.64 Relatively simple levels of theory (AM1) can efficiently model this arc-shaped topology63 giving the molecular architect significant control over final scaffold dimensions.
![Examples of [n]polynorbornane frameworks with curved and linear geometries as predicted by molecular modelling (r is the calculated radius of curvature).63](/image/article/2013/CC/c3cc40702k/c3cc40702k-f12.gif) | ||
| Fig. 12 Examples of [n]polynorbornane frameworks with curved and linear geometries as predicted by molecular modelling (r is the calculated radius of curvature).63 | ||
Fused [n]polynorbornanes and anion recognition65–69
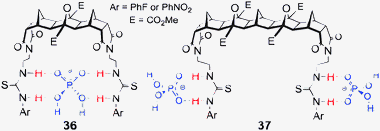
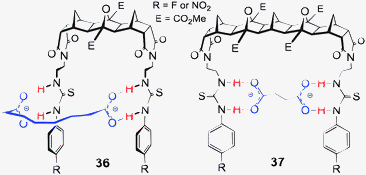
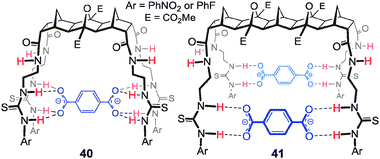
With the goal of creating a family of new hosts that could be used to target a range of larger and biologically relevant anions, such as dicarboxylates and pyrophosphate, a series of thiourea based anion receptors 36–41 (both symmetric and non-symmetric, Fig. 13) were designed. Using molecular modelling (AM1) it was calculated that the [3]polynorbornane 36 spans ca. 6.6 Å from imide N to imide N and the [5]polynorbornane 37 spans 10.4 Å.65 Thus the cleft dimensions of these polynorbornanes are significantly different and ideally suited to recognition of larger/longer anions. Again the 2-ureidoethylamido substituents are referred to as arms and as such the hosts will be referred to as 2, 3 or 4-armed [n]polynorbornanes, for example host 40a (Fig. 13) is a 4-armed [3]polynorbornane.![2,3 and 4-armed [3] and [5]polynorbornane hosts 36–41.67–69](/image/article/2013/CC/c3cc40702k/c3cc40702k-f13.gif) | ||
| Fig. 13 2,3 and 4-armed [3] and [5]polynorbornane hosts 36–41.67–69 | ||
Synthesis
Initially the desired 1- or 2-arm Boc protected norbornene unit was prepared. The protected [3]- or [5]polynorbornane framework was then assembled through the sequence of Mitsudo reaction, epoxidation, and ACE 1,3-dipolar cycloaddition. The final steps in all cases were deprotection then reaction with either 4-nitrophenyl- or 4-fluorophenylisothiocyanate. The two examples in Scheme 4 (2-armed receptor 36 and 4-armed receptor 41) illustrate the similar approaches.67–69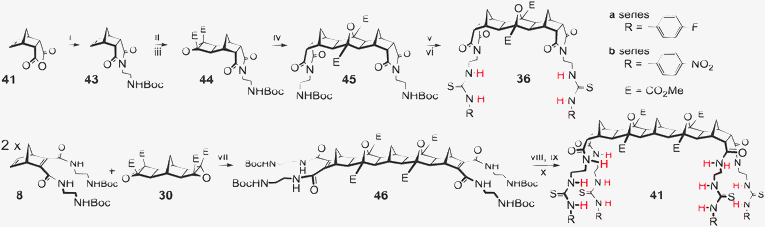 | ||
| Scheme 4 Synthesis of hosts 36 and 41. Reagents and conditions: (i) 2-(tert-butoxycarbonylamino)ethylamine, CHCl3, 120 °C, 12 h, 81% (ii) DMAD, RuH2(CO)(PPh3)3, THF, 70°C, 72 h, 86% (iii) TBHP, KOtBu, THF, 0 °C, 28 h, 69% (iv) DCM, 140 °C, 24 h, 58% (v) 20% TFA/CH2Cl2, 4 h, 100% (vi) DIPEA, CHCl3, 23 h, for 36a 4-fluorophenylisothiocyanate, 84%, for 36b 4-nitrophenylisothiocyanate, 68% (vii) 2.2 eq. 9, THF, 140 °C, 49 h, 65% (viii) H2, Pd–OH/C, 48 h 61% (ix) 20% TFA/CH2Cl2, 4 h, 100% (x) DIPEA, CHCl3, 24 h, for 41a 4-fluorophenylisothiocyanate, 92%, for 41b 4-nitrophenylisothiocyanate, 95% | ||
Construction of host 36 (Scheme 4) required norbornene imide 43 which was readily synthesised by heating anhydride 47 with 2-(tert-butoxycarbonylamino)ethylamine to give imide 48.67–69 The protocol of Mitsudo reaction with dimethylacetylene dicarboxylate (DMAD) followed by Weitz–Scheffer epoxidation gave oxirane 44. Subsequent ACE reaction of alkene 43 with cyclobutane epoxide 44 resulted in the Boc protected 2-armed [3]polynorbornane scaffold 45. Deprotection then coupling with the desired isothiocyanate gave hosts 36a and 36b.
For hosts 38–41 the previously synthesised norbornenes 4 and 5 and their precursors could be used as substrates for the construction of the 3- and 4-armed frameworks. Thus for the synthesis of 4-armed [5]polynorbornane host 41 norbornene 8 was employed, and in this case ACE reaction of bis-epoxide 30 with two equivalents of 8 provided the 4-armed [5]polynorbornane scaffold 46. Subsequent hydrogenation, deprotection and coupling with the requisite isothiocyanates afforded hosts 41a and 41b.
Anion binding studies
Two armed hosts: size matters65–67
As the new [3] and [5]polynorbornane hosts 36–41 have larger binding clefts than the simple norbornane based hosts 4–6, the set of anionic guests was also expanded to include alkyl dicarboxylates (−OOC(CH2)nCOO−, n = 1–6) and terephthalate; prepared as TBA salts.67 These anions were used in 1H NMR titration experiments in DMSO-d6 and selected results are summarised in Table 2.:G stoichiometries and calculated association constants (logK) for 2-arm hosts 36 and 37a
| 36a | 36b | 37a | 37b | ||
|---|---|---|---|---|---|
|
a Max Δδ obtained from ArN–H after addition of 5.0 eq. of anion; logK were determined by 1H NMR titration using WinEQNMR software20 (fittingprogram70 for terephthalate) with error ≤ 15%. Values for logK ≥ 5 are indicated as approximate as they are at the limits of accuracy for NMR. Titrations were carried out with [H]i of ∼1.2 × 10−2 M. D indicates deprotonation thus H:G stoichiometry and logK could not be determined.
|
|||||
| H2PO4− | max Δδ (ppm) | 2.0 | 1.9 | 2.5 | 2.3 |
| H:G |
1:1 |
1:1 |
1:2 |
1:2 |
|
| logK1 |
2.6 | 2.9 | 2.7 | 3.5 | |
| logK2 |
— | — | 2.5 | 3.0 | |
| AcO− | max Δδ (ppm) | 3.4 | 3.5 | 3.5 | 3.6 |
| H:G |
1:2 |
1:2 |
1:2 |
1:2 |
|
| logK1 |
2.8 | 3.2 | 2.9 | 3.1 | |
| logK2 |
2.3 | 3.0 | 2.3 | 3.0 | |
| Succinate2− (n = 2) | max Δδ (ppm) | 3.8 | 3.6 | 4.1 | 4.1 |
| H:G |
1:1 |
D | 1:1 |
1:1 |
|
| logK1 |
4.5 | — | 4.8 | ∼5.0 | |
| Suberate2− (n = 6) | max Δδ (ppm) | 3.7 | 3.9 | 3.9 | 4.0 |
| H:G |
1:1 |
1:1 |
1:1 |
1:1 |
|
| logK1 |
4.8 | ∼5.0 | ∼5.0 | ∼5.3 | |
| Terephthalate2− (n = phenyl) | max Δδ (ppm) | 3.38 | 3.51 | 3.64 | 3.74 |
| H:G |
1:1 |
1:1 |
1:1 |
1:1 |
|
| logK1 |
3.6 | 3.7 | 4.3 | ∼5.5 | |
Of the results obtained for the ‘smaller’ anions, the most useful were those obtained from the titrations against H2PO4−. For the [3]polynorbornane based host 36 1:1 H:G stoichiometry was observed in both cases, whereas [5]polynorbornane based hosts 37 formed 1:2 H:G complexes (Fig. 14). The H:G stoichiometry can be attributed to the shorter cleft width of host 36; the H2PO4− anion is simply too small to span the cleft of host 37 so cannot be bound cooperatively by the two anionophoric arms, instead each arm binds independently. This notion of the arms acting independently is further supported by the binding constants K1 and K2 being similar in magnitude (Table 2).
 | ||
| Fig. 14 Proposed binding conformations of host 36 binding one equivalent of H2PO4−, and host 37 in a 1:2 complex with H2PO4−. | ||
A trend was expected in which short chain dicarboxylates would complement the [3]polynorbornane host 36 and longer chain alkyl dicarboxylates would prefer the larger cleft of the [5]polynorbornane hosts 37. However, no such trend was found and in all cases strong 1:1 H:G complexes were observed (Table 2). In Fig. 15 two binding arrangements are shown; in the case of the [3]polynorbornane 36 binding suberate it is the flexibility of (i) the arms of the host and (ii) the alkyl chain of the guest that allows a conformation in which a strong host:guest complex forms. In the second example, despite the longer [5]polynorbornane scaffold of host 37 the flexibility of the arms still allows the host to capture the shorter succinate guest. The deliberate allowance for induced fit designed into these receptors was sufficient to over-ride the preorganisation imparted by the rigid scaffold.
 | ||
| Fig. 15 Proposed binding conformations of the 1:1 complexes formed between hosts 36 and 37 and the various length alkyl dicarboxylates. | ||
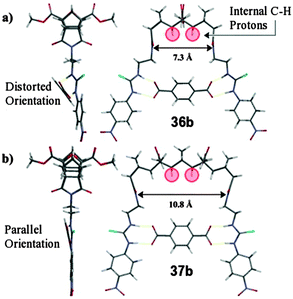 | ||
| Fig. 16 Molecular model calculated at H–F 3-21G* level of theory depicting the 1:1 complexes formed between the rigid aryl dicarboxylate, terephthalate2− and (a) host 36b and, (b) host 37b. Internal CH protons highlighted in red. | ||
The rigid terephthalate guest (7.0 Å long) can only bind strongly to a host with an appropriate cleft width and while a 1:1 H:G arrangement for both hosts 36 and 37 was identified there were significant differences between the titration isotherms and the binding constants. Host 37b bound terephthalate 100 times more strongly than host 36b.67 The cleft width of the host was now the controlling factor in the binding of the guest (illustrated in Fig. 16) where the larger cleft of host 37 better complements the width of the rigid dianionic guest. These results neatly reinforce the ideas of Cram who in his principle of preorganisation stated that “the more highly hosts and guests are organised for binding and low solvation prior to their complexation the more stable will be their complexes”.71
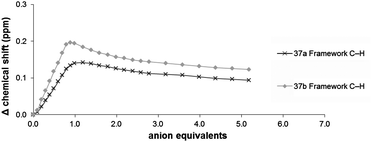
The interaction of host 37 with terephthalate could also be monitored by following the ‘internal’ framework C–H resonances as these protons are deshielded by the ring-current effect72 of the phenyl ring (Fig. 17).73 Although the observed change was small (Δδ ∼ 0.2 ppm) the binding isotherm clearly indicated the formation of a 1:1 complex (Fig. 17).
3 and 4 armed hosts: multiple H-bond donors68,69
The 3- and 4-armed [n]polynorbornane hosts 38 and 39 provide up to 12 H-bond donors per host (both thiourea and amide). As such they are ideally suited to larger guests with multiple H-bond acceptor sites and dihydrogenpyrophosphate (H2ppi2−) and adenosinediphosphate (ADP2−) were also included in the already large list of titrants for these hosts.69 Selected results are provided in Tables 3 (for 3-arm) and 4 (for 4-arm).:G stoichiometry and calculated association constants (logK) for 3-arm hosts 38 and 39a
| 38a | 38b | 39a | 39b | ||
|---|---|---|---|---|---|
|
a Max Δδ obtained from ArN–H after addition of 5.0 eq. of anion; logK were determined by 1H NMR titration using WinEQNMR software20 (fittingprogram70 for terephthalate) with (error ≤ 15%). Titrations were carried out with [H]i of ∼2.5 × 10−3 M. D indicates deprotonation therefore H:G stoichiometry and logK could not be determined. A indicates a high degree of aggregation was noted and assessment of the titration data was impossible. R indicates that binding was regioselective or occurred at one end of the framework only.
|
|||||
| H2PO4− | max Δδ (ppm) | 1.9 | 2.3 | 1.8 | 1.6 |
| H:G |
1:2R |
1:2R |
1:2R |
A | |
| logK1 |
3.5 | 2.7 | 3.3 | — | |
| logK2 |
<1 | <1 | <1 | — | |
| Pyrophosphate (H2ppi2−) | max Δδ (ppm) | ||||
| H:G |
1:1R |
1:1R |
1:1R |
A | |
| logK1 |
3.8 | 4.7 | 3.4 | — | |
| logK2 |
— | — | — | — | |
| AcO− | max Δδ (ppm) | 2.8 | 3.5 | 2.7 | 3.3 |
| H:G |
1:2 |
1:2 |
1:2 |
1:2 |
|
| logK1 |
2.9 | 3.2 | 2.9 | 3.1 | |
| logK2 |
2.5 | 2.5 | 2.4 | 2.6 | |
| Pimelate | max Δδ (ppm) | 3.5 | 3.2 | 3.4 | 3.2 |
| H:G |
1:2R |
1:1 |
1:2R |
1:1 |
|
| logK1 |
4.8 | 4.3 | 4.5 | 3.8 | |
| logK2 |
2.8 | D | 2.9 | D | |
| Terephthalate (n = aryl) | max Δδ (ppm) | 2.14 | 2.81 | 3.38 | 3.74 |
| H:G |
1:1 |
1:1 |
1:1 |
1:1 |
|
| logK1 |
3.0 | 3.2 | 4.4 | 4.9 | |
| logK2 |
— | — | — | — | |
Hosts 38 and 39 bound terephthalate in a 1:1 H:G arrangement and the guest was bound cooperatively through all six thiourea H-bond donors (no contribution from the amide groups). Due to the unsymmetrical nature of the 3-armed hosts, five H-bond donor signals could be followed throughout the 1H NMR titration (Fig. 18) and as such an increase in the amount of information regarding the binding could be gathered. A global method of calculating binding constants (taking into account all H-bond donors) could also be used to accurately determine logK.70
![Titration isotherm of host 39b upon the addition of terephthalate and proposed 1 : 1 complex formed between the 3 armed [5]polynorbornanes and terephthalate.](/image/article/2013/CC/c3cc40702k/c3cc40702k-f18.gif) | ||
| Fig. 18 Titration isotherm of host 39b upon the addition of terephthalate and proposed 1:1 complex formed between the 3 armed [5]polynorbornanes and terephthalate. | ||
:G stoichiometries and calculated association constants (logK) for 4-arm hosts 40–41a
| 40a | 40b | 41a | 41b | ||
|---|---|---|---|---|---|
|
a Max Δδ obtained from ArN–H after addition of 5.0 eq. of anion; logK were determined by 1H NMR titration using WinEQNMR software20 (fittingprogram70 for terephthalate) with (error ≤ 15%). Titrations were carried out with [H]i of ∼2.5 × 10−3 M.
|
|||||
| H2PO4− | max Δδ (ppm) | 1.8 | 1.8 | 1.6 | 2.1 |
| H:G |
1:2 |
1:2 |
1:2 |
1:2 |
|
| logK1 |
2.7 | 2.8 | 2.6 | 2.8 | |
| logK2 |
2.6 | 2.5 | 2.6 | 2.4 | |
| Pyrophosphate (H2ppi2−) | max Δδ (ppm) | 1.1 | 1.7 | 1.1 | 1.7 |
| H:G |
1:2 |
1:2 |
1:2 |
1:2 |
|
| logK1 |
3.0 | 3.0 | 4.2 | 2.4 | |
| logK2 |
2.2 | 3.0 | 2.5 | 1.7 | |
| AcO− | max Δδ (ppm) | 2.1 | 2.7 | 2.0 | 2.9 |
| H:G |
1:2 |
1:2 |
1:2 |
1:2 |
|
| logK1 |
2.8 | 2.9 | 2.7 | 3.0 | |
| logK2 |
2.4 | 2.7 | 2.5 | 2.6 | |
| Pimelate | max Δδ (ppm) | 3.1 | 3.2 | 3.0 | 3.2 |
| H:G |
1:2 |
1:2 |
1:2 |
1:2 |
|
| logK1 |
∼5.0 | ∼5.1 | 5.1 | ∼5.0 | |
| logK2 |
4.6 | 4.8 | 4.9 | ∼5.0 | |
| Terephthalate2− (n = aryl) | max Δδ (ppm) | 3.11 | 3.47 | 3.64 | 3.84 |
| H:G |
1:1 |
1:1 |
1:2 |
1:2 |
|
| logK1 |
2.9 | 3.0 | 4.1 | ∼5.0 | |
| logK2 |
— | — | 3.5 | 4.4 | |
It was also noted that the change in chemical shift of the thiourea protons of the one-armed end were approximately double that (at the equivalence point) of the shifts observed for the four thiourea H-bond donors of the 2-armed end. This result reinforces the idea that one carboxylate of the dianion is being bound by both thiourea groups from the 2-armed end of the host while the other carboxylate is being bound by the single thiourea group (Fig. 18).
Regioselective recognition69
The most remarkable behaviour for this series of hosts was observed when either alkyl dicarboxylates or pyrophosphate were added to the 3-armed hosts (38 and 39). A stepwise regioselective binding process occurred in the case of the alkyl dicarboxylates and in the case of pyrophosphate, a H:G complex formed in which the 1-armed end was completely ignored and the anion bound exclusively to the 2-armed end.
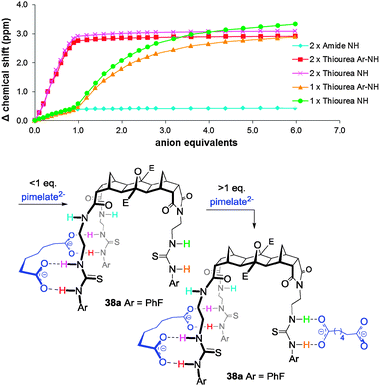
For the 3-armed hosts with alkyl dicarboxylates the clearest example of the stepwise binding was observed for host 38a with pimelate (Fig. 19). The binding isotherms indicated that an initial 1:1 binding event occurred at the 2-armed end. When one equivalent of dicarboxylate had been added there was little change in the urea protons of the 1-arm end (Fig. 19). When more than one equivalent of dicarboxylate was added no further change was observed at the 2-arm end, however, there was a distinct ‘jump’ in the N–H signals of the 1-arm end. The isotherm for the 1-armed end after one equivalent is reminiscent of a standard 1:1 binding isotherm and indicates modest binding of a second equivalent of pimelate at that end. Hence the overall process can be considered a stepwise regioselective process where the first dicarboxylate preferentially binds tightly at the 2-armed end.69 Modelling (H–F 3-21G*) also supported the binding of the dicarboxylate at the 2-arm end (Fig. 20).69
 | ||
| Fig. 19 Titration isotherm of host 38a with pimelate (above) and an illustration of the stepwise binding process. | ||
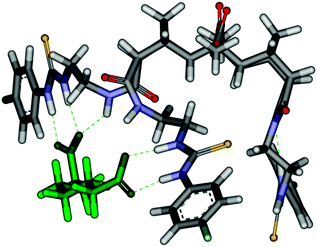 | ||
| Fig. 20 Molecular model calculated at Hartree–Fock 3-21G* level of theory depicting the 1:1 complex initially formed between host 38a and pimelate. | ||
To further demonstrate the regioselective binding of hosts 38 and 39, mixed anion titrations were conducted. By adding one equivalent of dicarboxylate (pimelate or malonate) followed by an excess of acetate is was possible to assemble in, a stepwise fashion, pimelate at the 2-armed end then acetate at the 1-arm end.
Our recent investigations into the binding of dihydrogenpyrophosphate (H2ppi2−) to the [n]polynorbornane hosts have used the tributylammonium salt [(Bu3NH)2H2ppi] and 3-arm hosts 38 and 39 both bound this form of H2ppi2− in a 1:1 H:G stoichiometry. The binding isotherms (Fig. 21) clearly indicate that the anion interacts with both the urea N−H protons as well as the amide N−H protons of the two-armed end whereas the single armed side appears to be completely ignored even when an excess of pyrophosphate was added. Regardless of the size of the binding cleft (either [3] or [5]polynorbornane) no response from the 1-arm end was noted. The cleft width also had little or no effect on the strength of the binding (Table 3). The contribution from the amide groups was considerable with the magnitude of the observed changes in chemical shift approaching that of the thiourea N−H (Fig. 21).
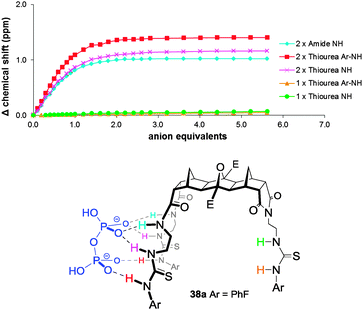 | ||
| Fig. 21 Titration isotherm of 3-arm host 38a upon the addition of H2ppi2− and illustration of the regioselective recognition of H2ppi2− by the 3-arm host 38. | ||
Due to the regioselective recognition ability of 38 and 39 with pyrophosphate and the stepwise assembly process previously accomplished using pimelate/acetate it was envisioned that with pyrophosphate bound at the 2-armed end, the single armed end should be free to bind a smaller anion such as phosphate or acetate. However, upon titration, the single armed end was still ignored, even when an excess of phosphate was added. Even when the anion addition order was reversed (one equivalent of phosphate was added followed by an excess of H2ppi2−) the isotherm quickly morphed into what would be expected of a pure H2ppi2− titration.
Both the size of the cleft and the urea electron withdrawing groups (Ar–F or Ar–NO2) had an influence on the ability of these hosts to regioselectively bind anions. The larger hosts (39) showed less selectivity than their smaller counterparts. The b-series (Ar–NO2) also showed less tendency towards regioselectivity than the a-series (Ar–F). While the exact cause of the regioselective binding remains unknown, one possibility, the formation of larger symmetric H:G complexes (e.g. 2:2), has been ruled out using NMR diffusion experiments.
One guest or two?
The last examples to be featured are those of the 4-arm hosts 40 and 41. In the titrations of dicarboxylates against these hosts a distinct ‘inflection’ in the binding isotherm appeared at approximately 1.0 eq. of anion (Fig. 22). It was reasoned that up until ca. 1.0 eq. of dicarboxylate had been added a 1:1 H:G arrangement was preferred then once an excess of anion was added a ‘switch’ to a 1:2 H:G stoichiometry occurred (Fig. 22).
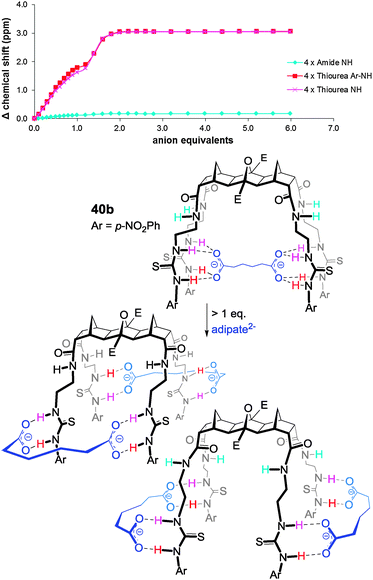 | ||
| Fig. 22 Isotherm and proposed binding of adipate by host 40b clearly showing the ‘switch’ from a 1:1 to a 1:2 H:G arrangement with an excess of guest. | ||
When titrations were performed using terephthalate and the 4-armed hosts a 1:1 H:G arrangement was noted for the [3]polynorbornane 40 but a 1:2 H:G complex for the longer [5]polynorbornane 41 (Fig. 23). Host 41 can encapsulate two rigid terephthalate dianions as the ethylene arms can flex away from the framework to minimise electrostatic repulsion between the two terephthalate units.
 | ||
| Fig. 23 Proposed 1:1 and 1:2 binding arrangements of hosts 40 and 41, respectively, when binding terephthalate. | ||
For H2ppi2− the titrations and Job plots showed that the 4-armed hosts 40 and 41 bind in a symmetric 1:2 H:G stoichiometry. The titrations identified that all 12 H-bond donors of the host were involved and the strength of the 1:2 complexes formed was independent of the cavity width of the host as evidenced by logK values The combination of results suggests that each side of the host was acting independently and the dianions are not spanning the cleft (Fig. 24).
 | ||
| Fig. 24 Proposed binding arrangement of host 41 (also representative of 40) with two equivalents of H2ppi2−. | ||
Conclusions and outlook
The outlook for further applications of norbornanes and [n]polynorbornanes in supramolecular chemistry is excellent as the full range of possible framework and cleft geometries is yet to be explored. The group at Deakin is currently developing multicomponent strategies for the rapid construction of [n]polynorbornane frameworks and aims to further expand the current range of hosts to include fluorescent signalling moieties and also to use these functionalised frameworks in applications such as organocatalysis.74 A full series of analogues of the LPS binder 10 are also in preparation. The results of these endeavours will be reported in due course.Acknowledgements
The authors thank the many excellent researchers in the field for continual inspiration. Particular thanks go to Professors Richard Russell and Thorfinnur Gunnlaugsson for introducing FP to BLOCK chemistry and anion recognition respectively.Notes and references
- Supramolecular Chemistry: from molecules to nanomaterials, ed. J. W. Steed and P. A. Gale, John Wiley and Sons, Chichester, 2012 Search PubMed; R. Vilar (Ed) Recognition of Anions, in series Structure and Bonding (series Ed. D. M. P. Mingos), Springer-Verlag, Berlin, 2008; J. L. Sessler, P. A. Gale and W.-S. Cho, Anion Receptor Chemistry, The Royal Society of Chemistry, Cambridge, UK, 2006 Search PubMed; J. L. Atwod and J. W. Steed, Encyclopedia of Supramolecular Chemistry, Marcel Dekker, New York, 2004 Search PubMed; A. Bianchi, K. Bowman-James and E. García-España, Supramolecular Chemistry of Anions, Wiley-VCH, New York, 1997 Search PubMed.
- For recent reviews see: M. Wenzel, J. R. Hiscock and P. A. Gale, Chem. Soc. Rev., 2012, 41, 480–520 RSC; A. E. Hargrove, S. Nieto, T. Z. Zhang, J. L. Sessler and E. V. Anslyn, Chem. Rev., 2011, 111, 6603–6782 CrossRef CAS; M. E. Moragues, R. Martinez-Manez and F. Sancenon, Chem. Soc. Rev., 2011, 40, 2593–2643 RSC; S. Kubik, Chem. Soc. Rev., 2010, 39, 3648–3663 RSC; C. Caltagirone and P. A. Gale, Chem. Soc. Rev., 2009, 38, 520–563 RSC; S. K. Kim, D. H. Lee, J. I. Hong and J. Yoon, Acc. Chem. Res., 2009, 42, 23–31 CrossRef; H. D. P. Ali, P. E. Kruger and T. Gunnlaugsson, New J. Chem., 2008, 32, 1153–1161 RSC; T. Gunnlaugsson, M. Glynn, G. M. Tocci, P. E. Kruger and F. M. Pfeffer, Coord. Chem. Rev., 2006, 250, 3094–3117 CrossRef.
- E. Fischer, Ber. Dtsch. Chem. Ges., 1890, 23, 2611 CrossRef CAS; E. Fischer, Ber. Dtsch. Chem. Ges., 1894, 27, 2985 CrossRef.
- D. E. Koshland Jr., Angew. Chem., Int. Ed., 1994, 33, 2375–2378 CrossRef; D. E. Koshland Jr., Proc. Natl. Acad. Sci. U. S. A., 1958, 44, 98 CrossRef.
- D. J. Cram, Science, 1988, 240, 760–767 CAS.
- C. Schmuck and V. Bickert, J. Org. Chem., 2007, 72, 6832–6839 CrossRef CAS; C. Schmuck and E. Dudaczek, Tetrahedron Lett., 2005, 46, 7101–7105 CrossRef; Y. H. Zhang, Z. M. Yin, Z. C. Li, J. Q. He and J. P. Cheng, Tetrahedron, 2007, 63, 7560–7564 CrossRef; P. A. Gale, Chem. Commun., 2005, 3761–3772 RSC; J. L. Sessler, N. M. Barkey, G. D. Pantos and V. M. Lynch, New J. Chem., 2007, 31, 646–654 RSC.
- C. Caltagirone, P. A. Gale, J. R. Hiscock, S. J. Brooks, M. B. Hursthouse and M. E. Light, Chem. Commun., 2008, 3007–3009 RSC; C. Caltagirone, J. R. Hiscock, M. B. Hursthouse, M. E. Light and P. A. Gale, Chem.–Eur. J., 2008, 14, 10236–10243 CrossRef CAS; F. M. Pfeffer, K. F. Lim and K. J. Sedgwick, Org. Biomol. Chem., 2007, 5, 1795–1799 Search PubMed; J. L. Sessler, D. G. Cho and V. Lynch, J. Am. Chem. Soc., 2006, 128, 16518–16519 CrossRef.
- S. M. Seo, E. J. Cho, S. J. Lee, K. C. Nam, S. H. Park and J. H. Jung, Microporous Mesoporous Mater., 2008, 114, 448–454 CrossRef CAS; H. A. Jeong, E. J. Cho, H. M. Yeo, B. J. Ryu and K. C. Nam, Bull. Korean Chem. Soc., 2007, 28, 851–854 CrossRef; E. J. Cho, J. W. Moon, S. W. Ko, J. Y. Lee, S. K. Kim, J. Yoon and K. C. Nam, J. Am. Chem. Soc., 2003, 125, 12376–12377 CrossRef; L. Sebo, B. Schweizer and F. Diederich, Helv. Chim. Acta, 2000, 83, 80–92 CrossRef.
- S. Banerjee, E. B. Veale, C. M. Phelan, S. A. Murphy, G. M. Tocci, L. J. Gillespie, D. O. Frimannsson, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2013, 42, 1601–1618 RSC; R. M. Duke, E. B. Veale, F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39, 3936–3953 RSC; E. B. Veale and T. Gunnlaugsson, J. Org. Chem., 2008, 73, 8073–8076 CrossRef CAS.
- S. J. Brooks, C. Caltagirone, A. J. Cossins, P. A. Gale and M. Light, Supramol. Chem., 2008, 20, 349–355 CrossRef CAS; S. K. Kim and J. Yoon, Chem. Commun., 2002, 770–771 RSC; J. L. Sessler, T. D. Mody, D. A. Ford and V. Lynch, Angew. Chem., Int. Ed. Engl., 1992, 31, 452–455 CrossRef; T. Gunnlaugsson, A. P. Davis, J. E. O'Brien and M. Glynn, Org. Lett., 2002, 4, 2449–2452 CrossRef.
- D. J. Mercer and S. J. Loeb, Chem. Soc. Rev., 2010, 39, 3612–3620 RSC; V. Amendola and L. Fabbrizzi, Chem. Commun., 2009, 513–531 RSC; J. W. Steed, Chem. Soc. Rev., 2009, 38, 506–519 RSC; J. Perez and L. Riera, Chem. Soc. Rev., 2008, 37, 2658–2667 RSC; A. J. Harte, P. Jensen, S. E. Plush, P. E. Kruger and T. Gunnlaugsson, Inorg. Chem., 2006, 45, 9465–9474 CrossRef CAS; S. Goetz and P. E. Kruger, Dalton Trans., 2006, 1277–1284 RSC; P. D. Beer, A. G. Cheetham, M. G. B. Drew, O. D. Fox, E. J. Hayes and T. D. Rolls, Dalton Trans., 2003, 603–611 RSC; J. V. Carolan, S. J. Butler and K. A. Jolliffe, J. Org. Chem., 2009, 74, 2992–2996 CrossRef.
- See: Merck Index, ed. M. J. O'Neil, 13th edn, 2001, Whitehouse Station, New Jersey Search PubMed.
- O. Diels and K. Alder, Justus Liebigs Ann. Chem., 1928, 460, 98–122 CrossRef CAS; O. Diels and K. Alder, Chem. Ber., 1929, 62, 2081–2097 CrossRef; L. R. Walters, in: Nobel Laureates in Chemistry 1901–1992, ed. L. K. James, American Chemical Society, Rahway, 1994, pp. 332–337 Search PubMed.
- For excellent reviews see: G. Buchbauer and I. Pauzenberger, Pharmazie, 1999, 54, 5–18 Search PubMed; G. Buchbauer, H. Spreitzer and H. Frei, Pharmazie, 1991, 46, 88–97 Search PubMed; G. Buchbauer, H. Spreitzer and H. Frei, Pharmazie, 1991, 46, 161–170 Search PubMed . For more recent examples see: D. W. Oliver and S. F. Malan, Med. Chem. Res., 2008, 137–151 CrossRef CAS; D. Rennison, S. Bova, M. Cavalli, F. Ricchelli, A. Zulian, B. Hopkins and M. A. Brimble, Bioorg. Med. Chem., 2007, 15(8), 2963–2974 CrossRef; A. J. Moreno-Vargas, C. Schutz, R. Scopelliti and P. Vogel, J. Org. Chem., 2003, 68, 5632–5640 CrossRef; L. Axford, J. R. Boot, T. M. Hotten, M. Keenan and F. M. Martin, et al. , Bioorg. Med. Chem. Lett., 2003, 13(19), 3277–3280 CrossRef; S. A. Hutchinson, S. P. Baker and P. J. Scammells, Bioorg. Med. Chem., 2002, 10, 1115–1122 CrossRef.
- I. Karle and D. Ranganathan, J. Mol. Struct., 2003, 85–96 CrossRef CAS; T. K. Chakraborty, P. Srinivasu, S. K. Kumar and A. C. Kunwar, J. Org. Chem., 2002, 67, 2093–2100 CrossRef; M. North, D. E. Hibbs, M. B. Hursthouse, I. G. Jones, W. Jones and A. Malik, J. Org. Chem., 1998, 63, 1496–1504 CrossRef.
- X. Verdaguer, J. Vazquez, G. Fuster, V. Bernardes-Genisson, A. E. Greene, A. Moyano, M. A. Pericas and A. Riera, J. Org. Chem., 1998, 63, 7037–7052 CrossRef CAS; A. Lattanzi, P. Iannece, A. Vicinanza and A. Scettri, Chem. Commun., 2003, 1440–1441 RSC; Y. Gnas and F. Glorius, Synthesis, 2006, 1899–1930 Search PubMed; P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2001, 40, 3726–3748 CrossRef; A. Piatek, C. Chapuis and J. Jurczak, Helv. Chim. Acta, 2002, 85, 1973–1988 CrossRef.
- T. Winkler, M. Bayrhuber, B. Sahlmann and R. Herges, Dalton Trans., 2012, 41, 7037–7040 RSC; T. Winkler, I. Dix, P. G. Jones and R. Herges, Angew. Chem., Int. Ed., 2003, 42, 3541–3544 CrossRef CAS.
- A. J. Lowe, G. A. Dyson and F. M. Pfeffer, Org. Biomol. Chem., 2007, 5, 1343–1346 CAS.
- A. J. Lowe, G. A. Dyson and F. M. Pfeffer, Eur. J. Org. Chem., 2008, 1559–1567 CrossRef CAS.
- M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311–312 RSC.
- T. Hayashita, T. Onodera, R. Kato, S. Nishizawa and N. Teramae, Chem. Commun., 2000, 755–756 RSC; T. Gunnlaugsson, A. P. Davis and M. Glynn, Chem. Commun., 2001, 2556–2557 RSC; T. Gunnlaugsson, A. P. Davis, J. E. O'Brien and M. Glynn, Org. Biomol. Chem., 2005, 3, 48–56 Search PubMed.
- C. R. H. Raetz and C. Whitfield, Annu. Rev. Biochem., 2002, 71, 635–700 CrossRef CAS; H. Nikaido, Microbiol. Mol. Biol. Rev., 2003, 67, 593–656 CrossRef.
- T. Velkov, P. E. Thomson, R. L. Nation and J. Li, J. Med. Chem., 2010, 53, 1898–1916 CrossRef CAS.
- P. Blondeau, M. Segura, R. Perez-Fernandez and J. de Mendoza, Chem. Soc. Rev., 2007, 36, 198–210 RSC; M. D. Best, S. L. Tobey and E. V. Anslyn, Coord. Chem. Rev., 2003, 240, 3–15 CrossRef CAS . For the synthesis of the di-boc aminoethylguandine used in the lipid A recognition work see: S. M. Hickey, T. D. Ashton, S. K. Khosa and F. M. Pfeffer, Synlett, 2012, 1779–1782 Search PubMed.
- L. C. Henderson, J. Li, R. L. Nation, T. Velkov and F. M. Pfeffer, Chem. Commun., 2010, 46, 3197–3199 RSC.
- M. Schnopp, S. Ernst and G. Haberhauer, Eur. J. Org. Chem., 2009, 213–222 CrossRef CAS; J. X. Lin, J. Lu, R. Cao, J. T. Chen and C. Y. Su, Dalton Trans., 2009, 1101–1103 RSC; P. A. Gale, S. E. Garcia-Garrido and J. Garric, Chem. Soc. Rev., 2008, 37, 151–190 RSC; M. Formica, V. Fusi, E. Macedi, P. Paoli, G. Piersanti, P. Rossi, G. Zappia and P. Orlando, New J. Chem., 2008, 32, 1204–1214 RSC; S. E. Garcia-Garrido, C. Caltagirone, M. E. Light and P. A. Gale, Chem. Commun., 2007, 1450–1452 RSC; K. A. Jolliffe, Supramol. Chem., 2005, 17, 81–86 CrossRef; H. J. Buschmann, E. Cleve and E. Schollmeyer, Inorg. Chem. Commun., 2005, 8, 125–127 CrossRef; K. H. Choi and A. D. Hamilton, J. Am. Chem. Soc., 2003, 125, 10241–10249 CrossRef.
- B. A. McNally, A. V. Koulov, T. N. Lambert, B. D. Smith, J. B. Joos, A. L. Sisson, J. P. Clare, V. Sgarlata, L. W. Judd, G. Magro and A. P. Davis, Chem.–Eur. J., 2008, 14, 9599–9606 CrossRef CAS; U. Maitra, Org. Biomol. Chem., 2008, 6, 657–669 Search PubMed; A. Kumar and P. S. Pandey, Org. Lett., 2008, 10, 165–168 CrossRef; A. P. Davis, Molecules, 2007, 12, 2106–2122 CrossRef; A. P. Davis, Coord. Chem. Rev., 2006, 250, 2939–2951 CrossRef; L. Fang, W. H. Chan, Y. B. He, D. W. J. Kwong and A. W. M. Lee, J. Org. Chem., 2005, 70, 7640–7646 CrossRef; S. Ghosh, A. R. Choudhury, T. N. G. Row and U. Maitra, Org. Lett., 2005, 7, 1441–1444 CrossRef; A. P. Davis and J. B. Joos, Coord. Chem. Rev., 2003, 240, 143–156 CrossRef.
- A. P. Davis, A. J. Ayling, M. N. Pérez-Payán and A. P. Davis, J. Am. Chem. Soc., 2001, 123, 12716–12717 CrossRef.
- S. D. Whitmarsh, A. P. Redmond, V. Sgarlata and A. P. Davis, Chem. Commun., 2008, 3669–3671 RSC.
- P. Prados and R. Quesada, Supramol. Chem., 2008, 20, 201–216 CrossRef CAS; P. K. Lo and M. S. Wong, Sensors, 2008, 8, 5313–5335 CrossRef; M. Hamon, M. Menand, S. Le Gac, M. Luhmer, V. Dalla and I. Jabin, J. Org. Chem., 2008, 73, 7067–7071 CrossRef; S. E. Matthews and P. D. Beer, Supramol. Chem., 2005, 17, 411–435 CrossRef; A. Casnati, F. Sansone and R. Ungaro, Acc. Chem. Res., 2003, 36, 246–254 CrossRef; J. L. Atwood and A. Szumna, Chem. Commun., 2003, 940–941 RSC.
- R. J. Scheerder, J. F. J. Engbersen, A. Casnati, R. Ungaro and D. N. Reinhoudt, J. Org. Chem., 1995, 60, 6448–6454 CrossRef.
- E. Quinlan, S. E. Matthews and T. Gunnlaugsson, J. Org. Chem., 2007, 72, 7497–7503 CrossRef CAS.
- O. A. Okunola, J. L. Seganish, K. J. Salimian, P. Y. Zavalij and J. T. Davis, Tetrahedron, 2007, 63, 10743–10750 CrossRef CAS; J. L. Seganish, P. V. Santacroce, K. J. Salimian, J. C. Fettinger, P. Zavalij and J. T. Davis, Angew. Chem., Int. Ed, 2006, 45, 3334–3338 CrossRef.
- P. A. Gale, Acc. Chem. Res., 2011, 44, 216–226 CrossRef CAS; C.-H. Lee, H. Miyaji, D.-W. Yoon and J. L. Sessler, Chem. Commun., 2008, 24–34 RSC; P. A. Gale, P. Anzenbacher and J. L. Sessler, Coord. Chem. Rev., 2001, 222, 57–102 CrossRef.
- H. Miyaji, S. J. Hong, S. D. Jeong, D. W. Yoon, H. K. Na, S. J. Hong, S. Ham, J. L. Sessler and C. H. Lee, Angew. Chem., Int. Ed., 2007, 47, 2508–2511 CrossRef.
- M. G. Fisher, P. A. Gale, J. R. Hiscock, M. B. Hursthouse, M. E. Light, F. P. Schmidtchen and C. C. Tong, Chem. Commun., 2009, 3017–3019 RSC.
- M. A. Palacios, R. Nishiyabu, M. Marquez and P. Anzenbacher, J. Am. Chem. Soc., 2007, 129, 7538–7544 CrossRef CAS.
- I. Marti, J. Rubio, M. Bolte, M. I. Burguete, C. Vicent, R. Quesada, I. Alfonso and S. V. Luis, Chem.–Eur. J., 2012, 18, 16728–16741 CrossRef CAS; C. E. Willans, S. French, K. M. Anderson, L. J. Barbour, J. A. Gertenbach, G. O. Lloyd, R. J. Dyer, P. C. Junk and J. W. Steed, Dalton Trans., 2011, 40, 573–582 RSC; M. Alajarin, R.-A. Orenes, J. W. Steed and A. Pastor, Chem. Commun., 2010, 46, 1394–1403 RSC; A. Barnard, S. J. Dickson, M. J. Paterson, A. M. Todd and J. W. Steed, Org. Biomol. Chem., 2009, 7, 1554–1561 Search PubMed; P. Piatek, Tetrahedron Lett., 2007, 48, 4427–4430 CrossRef; D. W. Yoon, S. D. Jeong, M. Y. Song and C. H. Lee, Supramol. Chem., 2007, 19, 265–270 CrossRef; D. R. Turner, M. J. Paterson and J. W. Steed, J. Org. Chem., 2006, 71, 1598–1608 CrossRef; B. T. Nguyen and E. V. Anslyn, Coord. Chem. Rev., 2006, 250, 3118–3127 CrossRef.
- N. L. Bill, D. S. Kim, S. K. Kim, J. S. Park, V. M. Lynch, N. J. Young, B. P. Hay, Y. J. Yang, E. V. Anslyn and J. L. Sessler, Supramol. Chem., 2012, 24, 72–76 CrossRef CAS.
- H. T. Ngo, X. J. Liu and K. A. Jolliffe, Chem. Soc. Rev., 2012, 41, 4928–4965 RSC; P. G. Young, J. K. Clegg, M. Bhadbhade and K. A. Jolliffe, Chem. Commun., 2011, 47, 463–465 RSC; S. J. Butler and K. A. Jolliffe, Org. Biomol. Chem., 2011, 9, 3471–3483 Search PubMed; M. J. McDonough, A. J. Reynolds, W. Y. G. Lee and K. A. Jolliffe, Chem. Commun., 2006, 2971–2973 RSC.
- P. G. Young and K. A. Jolliffe, Org. Biomol. Chem., 2012, 10, 2664–2672 CAS.
- K. P. McDonald, Y. R. Hua, S. Lee and A. H. Flood, Chem. Commun., 2012, 48, 5065–5075 RSC; Y. R. Hua and A. H. Flood, Chem. Soc. Rev., 2010, 39, 1262–1271 RSC; Y. J. Li and A. H. Flood, Angew. Chem., Int. Ed., 2008, 47, 2649–2652 CrossRef CAS.
- Y. Hua, R. O. Ramabhadran, J. A. Karty, K. Raghavachari and A. H. Flood, Chem. Commun., 2011, 47, 5979–5981 RSC; Y. Li and A. H. Flood, J. Am. Chem. Soc., 2008, 130, 12111–12122 CrossRef CAS.
- P. K. Eggers, N. Darwish, M. N. Paddon-Row and J. J. Gooding, J. Am. Chem. Soc., 2012, 134, 7539–7544 CrossRef CAS; N. Darwish, I. Diez-Perez, S. Y. Guo, N. J. Tao, J. J. Gooding and M. N. Paddon-Row, J. Phys. Chem. C, 2012, 116, 21093–21097 Search PubMed; P. K. Eggers, P. Da Silva, N. A. Darwish, Y. Zhang, Y. J. Tong, S. Ye, M. N. Paddon-Row and J. J. Gooding, Langmuir, 2010, 26, 15665–15670 CrossRef; M. Golic, M. R. Johnston, D. Margetic, A. C. Schultz and R. N. Warrener, Aust. J. Chem., 2006, 59, 899–914 CrossRef; M. R. Johnston and D. M. Lyons, Supramol. Chem., 2005, 17, 503–511 CrossRef; M. R. Johnston, M. J. Latter and R. N. Warrener, Aust. J. Chem., 2001, 54, 633–636 CrossRef; R. W. Warrener, D. Margetic, A. S. Amarasekara, D. N. Butler, I. B. Mahadaban and R. A. Russell, Org. Lett., 1999, 1, 199–202 CrossRef; K. A. Jolliffe, T. D. M. Bell, K. P. Ghiggino, S. J. Langford and M. N. Paddon-Row, Angew. Chem., Int. Ed., 1998, 37, 916–919 Search PubMed; M. N. Paddon-Row, Acc. Chem. Res., 1994, 27, 18–25 CrossRef.
- N. Darwish, I. Diez-Perez, P. Da Silva, N. J. Tao, J. J. Gooding and M. N. Paddon-Row, Angew. Chem., Int. Ed., 2012, 51, 3203–3206 CrossRef CAS.
- L. D. Van Vliet, T. Ellis, P. J. Foley, L. G. Liu, F. M. Pfeffer, R. A. Russell, R. N. Warrener, F. Hollfelder and M. J. Waring, J. Med. Chem., 2007, 50, 2326–2340 CrossRef CAS.
- S. P. Gaynor, M. J. Gunter, M. R. Johnston and R. N. Warrener, Org. Biomol. Chem., 2006, 4, 2253–2266 CAS.
- J. P. Mathias and F. Stoddart, Chem. Rev., 1992, 215–225 CAS.
- P. R. Ashton, G. R. Brown, N. S. Isaacs, D. Giuffrida, F. H. Kohnke, J. P. Mathias, A. M. Z. Slawin, D. R. Smith, J. F. Stoddart and D. J. Williams, J. Am. Chem. Soc., 1992, 114, 6330–6353 CrossRef CAS.
- F. G. Klarner and T. Schrader, Acc. Chem. Res., 2012 DOI:10.1021/ar300061c; B. Branchi, G. Bergamini, L. Fiandro, P. Ceroni, A. Alvino, G. Doddi, F. Vogtle and F. G. Klarner, Dalton Trans., 2011, 40, 1356–1364 RSC; B. Branchi, P. Ceroni, V. Balzani, M. C. Cartagena, F. G. Klarner, T. Schrader and F. Vogtle, New J. Chem., 2009, 33, 397–407 RSC; P. Talbiersky, F. Bastkowski, F. G. Klarner and T. Schrader, J. Am. Chem. Soc., 2008, 130, 9824–9828 CrossRef CAS; M. Lobert, H. Bandmann, U. Burkert, U. P. Buchele, V. Podsadlowski and F. G. Klarner, Chem.–Eur. J., 2006, 12, 1629–1641 CrossRef; F. G. Klarner, B. Kahlert, A. Nellesen, J. Zienau, C. Ochsenfeld and T. Schrader, J. Am. Chem. Soc., 2006, 128, 4831–4841 CrossRef; F. G. Klarner and B. Kahlert, Acc. Chem. Res., 2003, 36, 919–932 CrossRef.
- S. Sinha, Z. M. Du, P. Maiti, F. G. Klarner, T. Schrader, C. Y. Wang and G. Bitan, ACS Chem. Neurosci., 2012, 3, 451–458 CrossRef CAS; S. Sinha, D. H. J. Lopes, Z. M. Du, E. S. Pang, A. Shanmugam, A. Lomakin, P. Talbiersky, A. Tennstaedt, K. McDaniel, R. Bakshi, P. Y. Kuo, M. Ehrmann, G. B. Benedek, J. A. Loo, F. G. Klarner, T. Schrader, C. Y. Wang and G. Bitan, J. Am. Chem. Soc., 2011, 133, 16958–16969 CrossRef.
- S. Freye, J. Hey, A. Torras-Galan, D. Stalke, R. Herbst-Irmer, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2012, 51, 2191–2194 CrossRef CAS; D. M. Engelhard, S. Freye, K. Grohe, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2012, 51, 4747–4750 CrossRef; G. H. Clever, W. Kawamura, S. Tashiro, M. Shiro and M. Shionoya, Angew. Chem., Int. Ed., 2012, 51, 2606–2609 CrossRef.
- G. H. Clever, S. Tashiro and M. Shionoya, Angew. Chem., Int. Ed., 2009, 48, 7010–7012 CrossRef CAS.
- D. Margetic, P. Troselj and M. R. Johnston, Mini-Rev. Org. Chem., 2011, 8, 49–65 CrossRef CAS; G. Seitz and C. H. Gerninghaus, Pharmazie, 1994, 49, 102–106 Search PubMed; R. N. Warrener, D. N. Butler, W. Y. Liao, I. G. Pitt and R. A. Russell, Tetrahedron Lett., 1991, 32, 1889–1892 CrossRef; V. G. Seitz and H. Wassmuth, Chem.-Ztg., 1988, 112, 80–81 Search PubMed; F. Thalhammer, U. Wallfahrer and J. Sauer, Tetrahedron Lett., 1988, 29, 3231–3234 CrossRef.
- R. W. Warrener, D. N. Butler and R. A. Russell, Synlett, 1998, 566–573 CrossRef CAS.
- Many 1,3 dipolar cycloadditions are now referred to as ‘click’ reactions to reinforce the idea that reaction substrates are joined together in high yield and with no atom waste. See: H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- R. N. Warrener, A. C. Schultz, D. N. Butler, S. D. Wang, I. B. Mahadeban and R. A. Russell, Chem. Commun., 1997, 1023–1024 RSC.
- R. C. Foitzik, A. J. Lowe and F. M. Pfeffer, Tetrahedron Lett., 2009, 50, 2583–2584 CrossRef CAS.
- T. Mitsudo, T. Shinsugi, Y. Nakagawa and Y. J. Takegami, J. Org. Chem., 1979, 44, 4492 CrossRef CAS; T. Mitsudo, T. Kondo, Y. Ozaki and Y. Watanabe, Angew. Chem., Int. Ed. Engl., 1994, 33, 580–581 CrossRef; T. Mitsudo and T. Kondo, Proc. Jpn. Acad., Ser. B, 2007, 83, 65–76 CrossRef.
- M. D. Johnstone, A. J. Lowe, L. C. Henderson and F. M. Pfeffer, Tetrahedron Lett., 2010, 5889–5891 CrossRef CAS.
- E. Weitz and A. Scheffer, Ber. Dtsch. Chem. Ges., 1921, 54, 2327 CrossRef CAS; C. Clark, P. Hermans, O. Meth-Cohn, C. Moore, H. C. Taljaard and G. van Vuuren, Chem. Commun., 1986, 1378–1380 RSC.
- F. M. Pfeffer and R. A. Russell, J. Chem. Soc., Perkin Trans. 1, 2002, 2680–2685 RSC.
- R. N. Warrener, D. Margetic, G. Sun, A. S. Amarasekara, P. Foley, D. N. Butler and R. A. Russell, Tetrahedron Lett., 1999, 40, 4111–4114 CrossRef CAS.
- M. Golic, M. R. Johnston, D. Margetic, A. C. Schultz and R. N. Warrener, Aust. J. Chem., 2006, 59, 899–914 CrossRef CAS; M. R. Johnston, M. J. Gunter and R. N. Warrener, Tetrahedron, 2002, 58, 3445–3451 CrossRef; D. Margetić, M. R. Johnston, E. R. T. Tiekink and R. N. Warrener, Tetrahedron Lett., 1998, 39, 5277–5280 CrossRef.
- F. M. Pfeffer, T. Gunnlaugsson, P. Jensen and P. E. Kruger, Org. Lett., 2005, 7, 5357–5360 CrossRef CAS.
- F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Org. Biomol. Chem., 2007, 5, 1894–1902 CAS.
- A. J. Lowe and F. M. Pfeffer, Chem. Commun., 2008, 1871–1873 RSC.
- A. J. Lowe and F. M. Pfeffer, Org. Biomol. Chem., 2009, 7, 4233–4240 CAS.
- A. J. Lowe, B. M. Long and F. M. Pfeffer, J. Org. Chem., 2012, 77, 8507–8517 CrossRef CAS.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC; A. J. Lowe, F. M. Pfeffer and P. Thordarson, Supramol. Chem., 2012, 24, 585–594 CrossRef CAS.
- D. Cram, Angew. Chem., Int. Ed., 1988, 27, 1009–1020 CrossRef.
- G. C. Bassler, R. M. Silverstein and T. C. Morrill, Spectrometric Identification of Organic Compounds, John Wiley & Sons, Inc., New York, 4th edn, 1981 Search PubMed.
- For an example of the geometry required for an upfield shift see: D. N. Butler, M. H. Shang and R. N. Warrener, Tetrahedron Lett., 2000, 41, 5985–5989 CrossRef CAS.
- Z. G. Zhang and P. R. Schreiner, Chem. Soc. Rev., 2009, 38, 1187–1198 RSC.
Footnote |
| † Present address: Digital Biology Center, Bio-Rad Laboratories Pty., Ltd., Pleasanton, CA 94566, USA. |
| This journal is © The Royal Society of Chemistry 2013 |