 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Density functional theory calculations of XPS binding energy shift for nitrogen-containing graphene-like structures
K.
Artyushkova
*a,
B.
Kiefer
b,
B.
Halevi
a,
A.
Knop-Gericke
c,
R.
Schlogl
c and
P.
Atanassov
*a
aChemical and Nuclear Engineering Department, University of New Mexico, Albuquerque, NM 87131, USA. E-mail: kartyush@unm.edu; plamen@unm.edu
bPhysics Department, New Mexico State University, Las Cruzes, NM 88003, USA. E-mail: bkiefer@nmsu.edu
cFritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, 14195 Berlin, Germany. E-mail: knop@fhi-berlin.mpg.de; rs01@fhi-berlin.mpg.de
First published on 11th February 2013
Abstract
Our results validate the use of independent DFT predicted BE shifts for defect identification and constraining ambient pressure XPS observations for Me–Nx moieties in pyrolyzed carbon based ORR electrocatalysts. This supports the understanding of such catalysts as vacancy-and-substitution defects in a graphene-like matrix.
The active site/sites of non-Platinum Group Metal (non-PGM) oxygen reduction reaction (ORR) electrocatalysts have been contentious for the past 50 years.1–8 The continued interest in pyrolyzed macrocycles (porphyrins or phthalocyanines) or other nitrogen-containing electrocatalysts is largely motivated by the need to find cost-effective and efficient material solutions for replacing Pt in polymer electrolyte membrane (PEM) fuel cells.1,2,5,9–15 X-ray Photoelectron Spectroscopy (XPS) has been the main surface analysis method for determining the chemical environment and coordination of nitrogen and transition metal (Me) in the electrocatalyst.1,3,5,6,9,13,15–20 Even though there is an agreement that Me–Nx may serve as one of the possible active sites in ORR, the distribution of Me–N2vs. Me–N4 centers and their specific role still remain unresolved. XPS which heavily relies on use of reference spectra in accurate identification of species cannot address this issue directly as no reference compounds with Me–N2 moieties are available. Identification of the chemical species from XPS spectra generally involves peak deconvolution through curve-fitting. The assignment of peaks and nitrogen coordination are not straightforward due to overlapping peaks that appear within a narrow energy window of 2.5 eV and the “full-width-half-maximum” (fwhm) for individual species is on the order of 1.2–1.5 eV.21–24 Moreover, the majority of XPS studies use internal carbon for charge calibration of the spectra, while C 1s may have maximum at various binding energies (BEs), from 284 to 285.6 eV, depending on relative amounts of carbides, graphitic, aliphatic and oxygenated types of carbon present. Using C 1s as internal calibration standard may result in as large as 1 eV difference in calibration shift as compared to using Au 4f. The difference between positions of pyrrolic and pyridinic N is also about 1 eV. Using C as internal calibration standard, thus, may cause misidentification of N species and thus, may result in significant differences in the derived relative distribution of the pyrrolic vs. pyridinic type of N.
In order to address all of these complicating issues in obtaining accurate information on N speciation in Me–nitrogen–carbon (Me–N–C) oxygen reduction catalysts, we are employing (1) accurate charge correction of the spectra using a Au 4f line for each individual sample analysed, (2) DFT calculations of binding energy shifts of N 1s spectra, (3) comparison and evaluation of DFT predicted BE shifts to experimentally curve-fitted spectra of model compounds and finally (4) using the information obtained from DFT calculations as input for curve-fitting of in situ XPS spectra for distinguishing Me–N2versus Me–N4 species in reducing versus oxidizing atmospheres.
All catalysts presented in this work were synthesized using a modified sacrificial silica support method.18 Wet impregnation of precursors (such as H2 and Co porphyrins and Co phenanthroline) onto the surface of fumed silica (Cab-O-Sil TM EH-5, surface area: ∼400 m2 g−1) was implemented. The solid was ground to a fine powder and subjected to heat treatment in a N2 atmosphere at 800 °C for 3 h. Finally, the silica was leached out by means of an excess amount of 20 wt% of HF for 24 h and the resulting powder was washed with DI water until neutral reaction.
Conventional XPS spectra were acquired on a Kratos Axis Ultra spectrometer using a Al Kα monochromatic source. 99% pure Au powder was mounted on each sample and the Au 4f spectrum was used for charge calibration of spectra. Ambient pressure XPS experiments were carried out at ISISS (Innovative Station for In Situ Spectroscopy), the catalysis beamline of the Fritz Haber Institute at the 3rd generation synchrotron BESSY II (Helmholtz-Zentrum Berlin). The sample was mounted, chamber evacuated, heated to 60 °C, and survey and detailed scans were first acquired. The sample was then exposed to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 O2:H2O at 0.5 mbar and further high resolution scans were acquired. Ambient pressure X-ray photoelectron spectra were calibrated by the Fermi edge acquired for each element.
:4 O2:H2O at 0.5 mbar and further high resolution scans were acquired. Ambient pressure X-ray photoelectron spectra were calibrated by the Fermi edge acquired for each element.
Fig. 1 shows selected N 1s XPS spectra while Table 1 shows N 1s speciation results for the set of standards and metal-free and metal-containing Me–N–C electrocatalysts acquired using a conventional UHV XPS instrument. The binding energies of pure pyridinic and pure pyrrolic peaks are well within agreement with databases, 398.8 eV and 400.7 eV, respectively.25 Pyrolyzed H2TPP, which is a metal-less porphyrin-based macrocycle, has dominating peaks due to pyrrolic N and pyridinic N, amine and graphitic N. Two-metal containing samples, one based on Co-porphyrin, CoTPP, and another based on pyrolization of a mixture of bipyridine (as a source of N and C) and Fe acetate (as a metal source) have a peak at 399.8 eV which can be identified as nitrogen associated with a metal. The experimental shift observed for nitrogen when it is bound to metal, thus, is ∼1 eV with respect to the position of N in a pyridinic environment (398.8 eV).
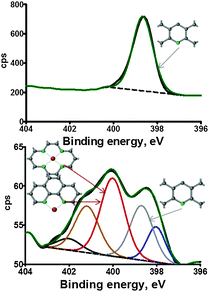 | ||
| Fig. 1 N 1s high resolution spectra of polypyridine (top) and bipyridine–Fe (bottom) catalysts. | ||
| Sample | Nitride | Pyride | Amine | N–Me | Pyrrolic | Graphitic-N–O |
|---|---|---|---|---|---|---|
| BE, eV | 398.0 | 398.8 | 399.3 | 399.8 | 400.7 | 402.3 |
| Poly-pyridine | — | 100 | — | — | — | — |
| Poly-pyrrole | — | — | 4.6 | — | 83.5 | 11.9 |
| H2TPP | 15.0 | 13.8 | 17.5 | — | 41.6 | 12.2 |
| CoTPP | 12.2 | 36.1 | — | 29.3 | 13.3 | 9.2 |
| Bipyridine–Fe | 12.5 | 22.5 | — | 36.4 | 21.4 | 7.2 |
There are only a few studies reporting density-functional-theory (DFT) calculations of BEs due to challenges that are associated with the treatment of core electrons and the steep and poorly screened Coulomb potential near the nucleus.26,27 Furthermore, pseudopotential based approaches dispense the core electrons and no knowledge of the inner electrons is usually retained. In contrast all-electron like PAW potentials retain orbital information of the core electrons, which is needed to compute properties such as the BE's of core electrons. While PAW potentials at least contain information about the core electrons the steep Coulomb potential remains a challenge. Our proposed solution to these challenges is to compare BE in the system of interest to a reference system that is accessible both in experiment and theory. Therefore we focus on BE shifts rather than BE's themselves. DFT calculations are being carried out as periodical solutions on graphene-like structures (infinite 2D C sheets), in which N and Me are introduced as substitution defects (see Fig. 2).
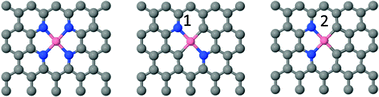 | ||
| Fig. 2 Structures as used for DFT calculations. | ||
All DFT computations were carried out using VASP28 at the GGA level using a periodic 3-d planewave simulation cell. The carbon support was modelled as a graphene sheet that contained 32 atoms. Interactions between valence electrons and nuclei are described with all-electron like PAW potentials.29 Spurious interactions between the modelled sheet and its periodic images perpendicular to the sheet were eliminated by introducing a 14 Å thick vacuum layer and by applying a dipole correction to the total energy. Core electron binding energies were calculated for the N 1s core state in the final state approximation. In order to obtain converged BEs from the computations, successively larger k-point grids up to 12 × 12 × 1 were chosen until converged BE differences between a reference material and the system of interest were obtained. The binding energy was determined as BE = BE(N 1s) − EF, where BE(N 1s) is the ab initio computed BE and EF is the corresponding Fermi energy.
Table 2 reports values of BE shift calculated for N in Co environments as shown in Fig. 2 and compares it with N 1s experimental values in Table 1. The average experimental BE shift reported for nitrogen bound to metal is very close to shifts calculated for three types of N environments as shown in Fig. 2.
| Type of bond | Pyridinic | Me–N4 | Me–N2 (1) | Me–N2 (2) |
|---|---|---|---|---|
| BE, eV | ΔE, eV | |||
| DFT shift | — | +1.1 | +0.8 | +1.0 |
| Experimental | 399.8 | +0.9–1.1 |
Peaks due to Me–N2 and Me–N4 will result in significant overlap as the difference in their position with respect to the position of pyridinic N is only 0.3 eV. Spectra acquired using synchrotron sources, however, have sufficient spectral resolution to deconvolve such closely overlapped peaks. Information obtained from DFT calculations can be used as input for determining BE shifts. Moreover, ambient pressure XPS using a synchrotron source allows for in situ analysis of electrocatalysts exposed to an oxidizing atmosphere at high temperatures providing information that is critical for identifying electro-catalytically active defect motifs.
Fig. 3 shows APXPS spectra of Co–phenanthroline electrocatalysts acquired under vacuum (a) versus those in an oxygen–water atmosphere at 60 °C (b). The spectra were curve fitted by fixing the positions of Co–N4 and Co–N2 with respect to pyridinic N as calculated by DFT (Table 2) which is 1.1 eV and 0.8 eV, respectively. The rest of the positions of pyrrolic and graphitic N were constrained to positions as determined in Fig. 1. It is obvious that the largest changes in the spectra when transferred from a reducing to an oxidizing atmosphere are in the range of BE's where nitrogen is bound to metal. The ratio of Co–N4/Co–N2 changes from 0.5 under vacuum to 2 in the presence of O2/H2O. This indicates that there is rearrangement of Co–Nx centers when material is being exposed to an oxidizing atmosphere. It may indicate that the presence of water and/or oxygen stabilizes Co–N4 centers. DFT calculations have indeed shown that Co–N4 centers are energetically more favorable for binding of oxygen than Co–N2 centers.30
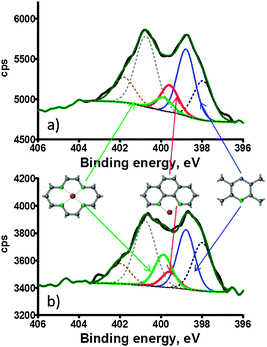 | ||
| Fig. 3 APXPS high resolution N 1s spectra of Co–phenanthroline acquired under (a) vacuum and (b) in O2/H2O at 60 °C. | ||
Our results validate for the first time the use of independent DFT predicted BE shifts for defect identification and constraining XPS observations for Me–Nx moieties in pyrolyzed carbon based ORR electrocatalysts. This supports the understanding of such catalysts as vacancy-and-substitution defects in a graphene-like matrix. Through careful spectra calibration and accurate DFT calculations of BE shifts for Me–nitrogen it is possible to highly reduce or eliminate the effect of the steep Coulomb potential on BE shifts. Therefore the synergy of experiment and theory allows extracting critical information from in situ high resolution N 1s spectra, which is needed for the determination of geometry and abundance of ORR active sites in an oxidizing atmosphere, which is critical information that must be evaluated against electrochemical activity for ORRs.
Notes and references
- K. Artyushkova, S. Pylypenko, T. S. Olson, J. E. Fulghum and P. Atanassov, Langmuir, 2008, 24, 9082–9088 CrossRef CAS.
- R. Bashyam and P. Zelenay, Nature, 2006, 443, 63–66 CrossRef CAS.
- M. Ferrandon, A. J. Kropf, D. J. Myers, K. Artyushkova, U. Kramm, P. Bogdanoff, G. Wu, C. M. Johnston and P. Zelenay, J. Phys. Chem. C, 2012, 116, 16001–16013 CAS.
- P. Gouerec and M. Savy, Electrochim. Acta, 1999, 44, 2653–2661 CrossRef CAS.
- M. Lefevre, E. Proietti, F. Jaouen and J. P. Dodelet, Science, 2009, 324, 71–74 CrossRef CAS.
- S. Pylypenko, A. Queen, T. S. Olson, A. Dameron, K. O'Neill, K. C. Neyerlin, B. Pivovar, H. N. Dinh, D. S. Ginley, T. Gennett and R. O'Hayre, J. Phys. Chem. C, 2011, 115, 13667–13675 CAS.
- L. T. Qu, Y. Liu, J. B. Baek and L. M. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS.
- Y. Y. Shao, J. H. Sui, G. P. Yin and Y. Z. Gao, Appl. Catal., B, 2008, 79, 89–99 CrossRef CAS.
- K. Artyushkova, S. Levendosky, P. Atanassov and J. Fulghum, Top. Catal., 2007, 46, 263–275 CrossRef CAS.
- E. J. Biddinger, D. von Deak and U. S. Ozkan, Top. Catal., 2009, 52, 1566–1574 CrossRef CAS.
- Z. W. Chen, D. Higgins, A. P. Yu, L. Zhang and J. J. Zhang, Energy Environ. Sci., 2011, 4, 3167–3192 CAS.
- F. Jaouen, E. Proietti, M. Lefevre, R. Chenitz, J. P. Dodelet, G. Wu, H. T. Chung, C. M. Johnston and P. Zelenay, Energy Environ. Sci., 2011, 4, 114–130 CAS.
- T. S. Olson, S. Pylypenko, J. E. Fulghum and P. Atanassov, J. Electrochem. Soc., 2010, 157, B54–B63 CrossRef CAS.
- J. D. Wiggins-Camacho and K. J. Stevenson, J. Phys. Chem. C, 2011, 115, 20002–20010 CAS.
- G. Wu, C. M. Johnston, N. H. Mack, K. Artyushkova, M. Ferrandon, M. Nelson, J. S. Lezama-Pacheco, S. D. Conradson, K. L. More, D. J. Myers and P. Zelenay, J. Mater. Chem., 2011, 21, 11392–11405 RSC.
- K. Artyushkova, D. Habel-Rodriguez, T. S. Olson and P. Atanassov, J. Power Sources, 2013, 226, 112–121 CrossRef CAS.
- A. Cimino, D. Gazzoli and M. Valigi, J. Electron Spectrosc. Relat. Phenom., 1999, 104, 1–29 CrossRef CAS.
- A. Serov, M. H. Robson, K. Artyushkova and P. Atanassov, Appl. Catal., B, 2012, 127, 300–306 CrossRef CAS.
- A. Serov, M. H. Robson, B. Halevi, K. Artyushkova and P. Atanassov, Electrochem. Commun., 2012, 22, 53–56 CrossRef CAS.
- A. M. Venezia, Catal. Today, 2003, 77, 359–370 CrossRef CAS.
- J. R. Pels, F. Kapteijn, J. A. Moulijn, Q. Zhu and K. M. Thomas, Carbon, 1995, 33, 1641–1653 CrossRef CAS.
- K. Stanczyk, R. Dziembaj, Z. Piwowarska and S. Witkowski, Carbon, 1995, 33, 1383–1392 CrossRef CAS.
- M. A. Wojtowicz, J. R. Pels and J. A. Moulijn, Fuel, 1995, 74, 507–516 CrossRef CAS.
- K. Stanczyk, Energy Fuels, 1999, 13, 82–87 CrossRef CAS.
- G. Beamson and S. Briggs, High Resolution XPS of Organic Polymers. The Scienta ESCA300 Database, Wiley & Sons, 1992 Search PubMed.
- D. P. Chong, J. Chem. Phys., 1995, 103, 1842–1845 CrossRef CAS.
- Y. Takahata and A. D. Marques, J. Electron Spectrosc. Relat. Phenom., 2010, 178, 80–87 CrossRef.
- G. Kresse and J. Hafner, J. Phys.: Condens. Matter, 1994, 6, 8245–8257 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B, 1999, 59, 1758–1775 CrossRef CAS.
- S. Kattel, P. Atanassov and B. Kiefer, Phys. Chem. Chem. Phys., 2013, 15, 148–153 RSC.
| This journal is © The Royal Society of Chemistry 2013 |