 Open Access Article
Open Access ArticleReduction of 2,2,2-trichloro-1-arylethanones by RMgX: mechanistic investigation and the synthesis of substituted α,α-dichloroketones†
Ali H.
Essa
ab,
Reinner I.
Lerrick
ac,
Floriana
Tuna
d,
Ross W.
Harrington
a,
William
Clegg
a and
Michael J.
Hall
*a
aSchool of Chemistry, Bedson Building, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: m.hall@ncl.ac.uk; Fax: +44 (0)191 222 6929; Tel: +44 (0)191 222 7321
bDepartment of Chemistry, College of Science, University of Basrah, Basrah, Iraq
cSchool of Chemistry, Nusa Cendana University, Indonesia
dSchool of Chemistry and Photon Science Institute, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 19th February 2013
Abstract
2,2,2-Trichloro-1-arylethanones undergo high yielding reductions to the corresponding 2,2-dichloro-1-arylethanones in the presence of RMgX. A single electron transfer mechanism for the reaction is proposed based on trapping experiments. Reaction of the intermediate enolates with a range of electrophiles is described, providing a convenient route to substituted α,α-dichloro-β-hydroxyketones and related molecules.
α,α-Dichlorocarbonyls are versatile synthetic intermediates, typically formed by α-chlorination of carbonyls,1 chlorination of silyl enolates,2 electrochemical or metal mediated reductions,3 aldol reactions4 or cycloadditions with dichloroketene.5 α,α-Dichlorocarbonyl groups have been employed in intramolecular radical cyclisations,6 have been converted to chloroalkenes,7 chlorooxiranes allowing access to α-keto esters8 and heteroaromatics,9 have been used as chlorinating agents10 and were found in the natural product chlorotonil A.11 In designing new routes to functionalised α,α-dichlorocarbonyls, we decided to investigate conditions for the reduction of 2,2,2-trichloro-1-arylethanones. We envisaged that 2,2,2-trichloro-1-arylethanones being sterically hindered and electron-deficient aromatic ketones, would form reactive ketyl radical anions in the presence of a suitable single electron donor such as a Grignard reagent.12 Further reaction of the intermediate ketyl radical anion would then provide a new route towards substituted α,α-dichloroketones.
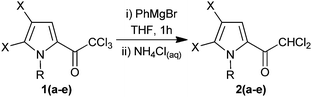
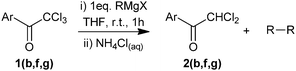
Our initial investigations involved the addition of commercially available 2,2,2-trichloro-1-(1H-pyrrol-2-yl)ethanone (1a) to PhMgBr, followed by quenching with excess aqueous NH4Cl. With the use of 1.1 equivalents of PhMgBr, at either 0 °C or r.t., the reaction resulted in the isolation of 2,2-dichloro-1-(1H-pyrrol-2-yl)ethanone (2a) in 50–55% yield (Table 1, entries 1 and 2), formally a C–Cl to C–H reduction.
| Entry | R | X | PhMgBr | Temp. | Product | Yieldb (%) |
|---|---|---|---|---|---|---|
| a The reactions were performed by reverse addition of 1 mmol of ketone in 1 mL of THF to a 2 M solution of PhMgBr in THF. b Isolated yields. | ||||||
| 1 | H | H | 1.1 eq. | 0 °C | 2a | 50 |
| 2 | H | H | 1.1 eq. | r.t. | 2a | 55 |
| 3 | H | H | 2.2 eq. | r.t. | 2a | 90 |
| 4 | Me | H | 1.0 eq. | r.t. | 2b | 94 |
| 5 | H | Cl | 2.0 eq. | r.t. | 2c | 87 |
| 6 | H | Br | 2.0 eq. | r.t. | 2d | 95 |
| 7 | H | I | 2.0 eq. | r.t. | 2e | 93 |
We postulated that the reaction may not go to completion due to competing deprotonation of the pyrrolic NH. Thus we re-examined the reaction of compound 1a with 2.2 equivalents PhMgBr at r.t., and the reaction of compound 1b (an analogous N-methylated compound) with 1.0 equivalents of PhMgBr (Table 1, entries 3 and 4). In both cases near-quantitative yields of the corresponding reduced compounds 2a and 2b were isolated after quenching of the reaction. In addition three di-halogenated pyrrole derivatives (1c–e) were also submitted to the optimised reaction conditions, again yielding the corresponding reduced products (2c–e) in high yield. Observation of this C–Cl to C–H reduction prompted us to investigate the mechanism in more detail.
After the reaction of 1a–e with PhMgBr and quenching with aqueous NH4Cl, a major by-product was observed by 1H NMR spectroscopy of the crude reaction mixtures, which on isolation by silica gel chromatography was identified as 1,1′-biphenyl. In the case of entry 3, 40 mg of 1,1′-biphenyl could be isolated, corresponding to approximately 50% of the total added PhMgBr. The formation of significant quantities of 1,1′-biphenyl is most likely to arise from the dimerisation of phenyl radicals generated during the reaction.13,14
To examine this supposition further we carried out an in situ EPR experiment. Solutions containing ketone 1b and PhMgBr in THF were added sequentially to an EPR tube cooled in liquid N2. Further addition of the spin trap 5,5-dimethyl-1-pyrroline N-oxide (DMPO) and subsequent warming to 270 K of the reaction mixture produced a species with a well-resolved, 6-line EPR spectrum (Fig. 1), corresponding to a Ph–DMPO adduct (2,2-dimethyl-5-phenylpyrrolidin-1-olate radical) with distinctive 1H and 14N hyperfine coupling constants: a(1H) = 20.3 G and a(14N) = 13.87 G. A minor product, the 2,2-dimethyl-5,5-diphenylpyrrolidin-1-olate radical, was also observed in the EPR spectrum (3 lines, a(14N) = 13.87 G). In the absence of ketone 1b neither adduct was observed, indicating that the Ph radical is generated under the reaction conditions.
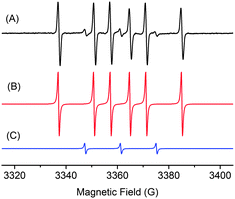 | ||
| Fig. 1 (A) X-band EPR spectrum at 270 K of a THF solution resulting from the reaction of PhMgBr with ketone 1b, in the presence of DMPO; (B) simulated spectrum of the Ph–DMPO adduct with the parameters: g = 2.0096; a(1H) = 20.3 G and a(14N) = 13.87 G; (C) simulated spectrum of the Ph2–DMPO adduct, with g = 2.0095 and a(14N) = 13.87 G. (100 kHz modulation frequency, 1 G modulation amplitude, 0.27 mW incident microwave power). | ||
To further probe 1,1′-biphenyl formation we examined the reaction between ketone 1b and a number of other RMgX species, where R was a para-substituted phenyl group (Table 2).
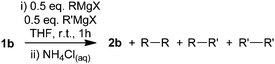
Reaction of 4-Me(C6H4)MgI with ketone 1b (Table 2, entry 1) gave 4,4′-dimethyl-1,1′-biphenyl as a single regioisomer. One to one mixtures of para-substituted phenyl Grignard regents (Table 2, entries 2–4) gave in each case a mixture of 4,4′-disubstituted-1,1′-biphenyl products. This suggests that the aryl–aryl bond is being formed at the position of the original C–Mg bond, supporting the formation 1,1′-biphenyl products via a radical coupling mechanism. This suggests that the RMgX is donating a single electron from the R–Mg bond to the substrate.
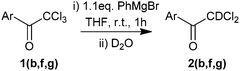
We postulated that the overall reaction involves a late stage enolate intermediate, which is quenched by aqueous NH4Cl to give the observed reduction product. To confirm this, ketones 1b, 1f and 1g were reacted with PhMgBr and quenched with D2O to give, in 50–96% yield, the corresponding deuterated products 2b, 2f, and 2g with high levels of D incorporation (Table 3).
We then investigated the influence of the R (aryl, alkyl) and X (halogen) groups of the Grignard reagent (Table 4).
| Entry | R | X | Ar | Yield/2a (%) | Yield/R2b (%) |
|---|---|---|---|---|---|
| a Isolated yields. b Yield based on total RMgX added. | |||||
| 1 | Et | Br | 1-Methyl-1H-pyrrol-2-yl (1b) | 50 | nd |
| 2 | i-Pr | Cl | 1-Methyl-1H-pyrrol-2-yl (1b) | 42 | nd |
| 3 | Ph | Cl | 1-Methyl-1H-pyrrol-2-yl (1b) | 61 | 45 |
| 4 | Ph | Br | 1-Methyl-1H-pyrrol-2-yl (1b) | 94 | 52 |
| 5 | Ph | I | 1-Methyl-1H-pyrrol-2-yl (1b) | 94 | 62 |
| 6 | Et | Br | p-Tolyl (1f) | 33 | nd |
| 7 | i-Pr | Cl | p-Tolyl (1f) | 33 | nd |
| 8 | Ph | Cl | p-Tolyl (1f) | 47 | 35 |
| 9 | Ph | Br | p-Tolyl (1f) | 68 | 38 |
| 10 | Ph | I | p-Tolyl (1f) | 96 | 71 |
| 11 | Ph | Br | 1-(4-(tert-Butyl)phenyl) (1g) | 71 | 39 |
| 12 | Ph | I | 1-(4-(tert-Butyl)phenyl) (1g) | 98 | 58 |
A comparison of Et, i-Pr and Ph groups (Table 4) showed that the highest yields resulted from the use of aryl Grignards.15 In addition, reaction yields showed the trend: X = I > Br > Cl.
Variation of solvent (THF, Et2O and hexane) and concentration had little effect on reaction outcomes. Only with extreme dilution was any influence noticeable (see ESI†).
Therefore we propose a potential mechanism for the Grignard-mediated reduction of trichloroacetyl-substituted aromatics. We suggest that the first step of the reaction is a single electron transfer from the Grignard reagent to the ketone. This intermediate radical anion then either: (a) loses a chlorine atom, (b) accepts a second electron and subsequently loses chloride or (c) loses chloride followed by addition of a second electron, to give the corresponding magnesium enolate (Fig. 2).16
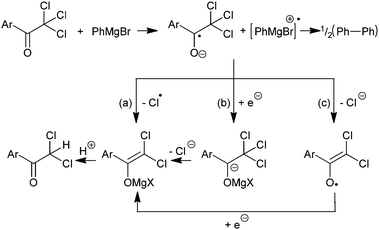 | ||
| Fig. 2 Proposed reaction pathways. | ||
Since the intermediate magnesium enolates can be intercepted by electrophiles, we have exploited this chemistry as a convenient “one-pot” reductive-functionalisation of 2,2,2-trichloro-1-(1-methyl-1H-pyrrol-2-yl)ethanone (1b) to give substituted α,α-dichloroketones. Reaction with 1-(chloromethyl)-4-nitrobenzene gave only a moderate yield of the expected product. Good yields were however obtained on reaction with diethyl 2-oxomalonate, aryl acid chlorides or aryl aldehydes (Table 5).17
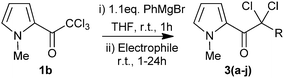
| Producta | Electrophile | R | Yieldb (%) |
|---|---|---|---|
| a 1b was reacted in THF with PhMgBr at r.t. for 1 h, after which a suitable electrophile was added and the mixture stirred at r.t. until TLC analysis showed that the reaction was complete. b Isolated yields. c Structures confirmed by single-crystal X-ray analysis. | |||
| 3a | PhCHO | PhCH(OH) | 81c |
| 3b | 4-MeO(C6H4)CHO | 4-MeO(C6H4)CH(OH) | 85c |
| 3c | 4-I(C6H4)CHO | 4-I(C6H4)CH(OH) | 94 |
| 3d | 5-Me(C4H2O)CHO | 5-Me(C4H2O)CH(OH) | 70 |
| 3e | C6F5CHO | C6F5CH(OH) | 70 |
| 3f | 4-NO2(C6H4)CHO | 4-NO2(C6H4)CH(OH) | 96c |
| 3g | 4-NO2(C6H4)CH2Cl | 4-NO2(C6H4)CH2 | 37c |
| 3h | 4-NO2(C6H4)COCl | 4-NO2(C6H4)C(O) | 95c |
| 3i | (EtO2C)2CO | (EtO2C)2C(OH) | 75 |
| 3j | C6H5COCl | C6H5C(O) | 50 |
In conclusion we have demonstrated a new approach to functionalised α,α-dichloroketones, via the reaction of commercially available RMgX reagents with 2,2,2-trichloro-1-aryl-ethanones. Additional examination of the substrate scope and investigations into subsequent synthetic modification of the α,α-dichloroketones formed will be discussed in future publications.
The authors thank the Iraqi Ministry of Education (A.H.E.) and the Indonesian Ministry of National Education (R.I.L.) for funding, ESPRC for X-ray facilities at Newcastle (EP/F03637X/1), the EPSRC National EPR Facility, the EPSRC National Mass Spectrometry Service, Prof. W. McFarlane and Dr C. Wills (NCL) for NMR support, and O. Aslan, M. Dunn, A. Nag and E. Çiftçi (NCL) for synthesis of 1d–g.
Notes and references
- F. Bellesia, F. Ghelfi, F. Reverberi, F. Danna, V. Frenna, F. Felluga, A. F. Parsons and D. Spinelli, Synthesis, 2012, 605 Search PubMed; Z. Chen, B. Zhou, H. Cai, W. Zhu and X. Zou, Green Chem., 2009, 11, 275 RSC; R. Akula, M. Galligan and H. Ibrahim, Chem. Commun., 2009, 6991 RSC; J.-J. Kim, D. H. Kweon, S.-D. Cho, H.-K. Kim, S.-G. Lee and Y.-J. Yoon, Synlett, 2006, 194 Search PubMed; R. V. Hoffman, W. S. Weiner and N. Maslouh, J. Org. Chem., 2001, 66, 5790 CrossRef CAS.
- Y. Zhang, K. Shibatomi and H. Yamamoto, J. Am. Chem. Soc., 2004, 126, 15038 CrossRef CAS.
- G. Quintanilla, I. Pérez, L. Záková, C. Uth and F. Barba, Eur. J. Org. Chem., 2011, 4681 CrossRef CAS; T. Imanishi, Y. Fujiwara, Y. Sawama, Y. Monguchi and H. Sajiki, Adv. Synth. Catal., 2012, 354, 771 CrossRef.
- H. Sasai, S. Arai and M. Shibasaki, J. Org. Chem., 1994, 59, 2661 CrossRef CAS; R. Imashiro and T. Kuroda, J. Org. Chem., 2003, 68, 974 CrossRef.
- C. Roche, K. Kadlečíková, A. Veyron, P. Delair, C. Philouze, A. E. Greene, D. Flot and M. Burghammer, J. Org. Chem., 2005, 70, 8352 CrossRef CAS.
- D. Yang, Y.-L. Yan, B.-F. Zheng, Q. Gao and N.-Y. Zhu, Org. Lett., 2006, 8, 5757 CrossRef CAS.
- Z. T. Narumi, T. Kobayakawa, H. Aikawa, S. Seike and H. Tamamura, Org. Lett., 2012, 14, 4490 CrossRef CAS; D. K. Barma, A. Kundu, H. Zhang, C. Mioskowski and J. R. Falck, J. Am. Chem. Soc., 2003, 125, 3218 CrossRef.
- D. Yang, M. Yang and N.-Y. Zhu, Org. Lett., 2003, 5, 3749 CrossRef CAS.
- G. R. Lawton, H. Ji, P. Martásek, L. J. Roman and R. B. Silverman, Beilstein J. Org. Chem., 2009, 5 CrossRef CAS , No. 28 F. Serra, P. Coutrot, M. Estève-Quelquejeu, P. Herson, T. K. Olszewski and C. Grison, Eur. J. Org. Chem., 2011, 1841 CrossRef.
- H. Wack, A. E. Taggi, A. M. Hafez, W. J. Drury III and T. Lectka, J. Am. Chem. Soc., 2001, 123, 1531 CrossRef CAS.
- N. Rahn and M. Kalesse, Angew. Chem., Int. Ed., 2008, 47, 597 CrossRef CAS.
- E. C. Ashby and A. B. Goel, J. Am. Chem. Soc., 1981, 103, 4983 CrossRef CAS; E. C. Ashby, Pure Appl. Chem., 1980, 52, 545 CrossRef; E. C. Ashby, Acc. Chem. Res., 1988, 414 CrossRef.
- Trace 2-phenyltetrahydrofuran could also be detected by 1H NMR and GCMS, when reactions were carried out in THF. We postulate that this is formed through radical H abstraction from the α-position of THF, followed by coupling of resulting THF derived radical with a phenyl radical.
- Commercial PhMgBr was shown to contain <2 mg of Ph2 per mmol.
- J. Villieras and B. Castro, Bull. Soc. Chim. Fr., 1970, 3, 1189 Search PubMed; K. Maruyama, Y. Matano and T. Katagiri, J. Phys. Org. Chem., 1991, 4, 501 CrossRef CAS.
- No evidence of atomic chlorine or bromine was observed by EPR. Neither C6H5Cl nor C6H5Br (the coupling products of phenyl radical and atomic halogen) could be detected by GCMS of the crude reaction mixture leading to 2b.
- The corresponding magnesium enolate can be prepared from 2b through deprotonation with NaH in THF, followed by ion exchange with MgCl2. The enolate was reacted with D2O to give a 78% yield (84% deuterium incorporation by 1H NMR) of (2-d)-2b or 4-NO2(C6H4)CHO to give a 47% yield of 3f.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, 1H and 13C spectra and X-ray structures. Crystallographic data for 1d, 3a, 3b, 3f, 3g and 3h, have been deposited with the CCDC, deposition nos: CCDC 916095–916100. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc39147g |
| This journal is © The Royal Society of Chemistry 2013 |