 Open Access Article
Open Access ArticleMonitoring fast reactions by spatially-selective and frequency-shifted continuous NMR spectroscopy: application to rapid-injection protein unfolding†
Gabriel E.
Wagner
a,
Peyman
Sakhaii
b,
Wolfgang
Bermel
c and
Klaus
Zangger
*a
aInstitute of Chemistry, University of Graz, Heinrichstrasse 28, 8010 Graz, Austria. E-mail: klaus.zangger@uni-graz.at
bNMR Laboratory of C&BD, Industriepark Hoechst, Building D770, Labor 204, 65926 Frankfurt/Main, Germany
cBruker BioSpin GmbH, 76287 Rheinstetten, Germany
First published on 1st March 2013
Abstract
The repetition rate of an NMR experiment is usually limited by the longitudinal relaxation times of the investigated molecule. Here we show that continuous excitation and data acquisition, without any interscan delay, is possible by a spatially resolved experiment where different nuclei are excited in consecutive scans.
NMR spectroscopy is a powerful and widely used method to study diverse molecular features under equilibrium conditions. The temporal resolution for the observation of irreversible reaction kinetics by NMR is quite restricted as the lowest achievable sampling rate is in the range of seconds. The reason for this limitation is the long longitudinal relaxation time (T1) of nuclear spins. Between successive scans the magnetization has to return close to its thermal equilibrium in order to avoid progressive saturation which would lead to intensity variations and in the worst case disappearance of signals. For exact intensity comparisons the relaxation delay between scans should be several times T1, which can be up to minutes for slowly relaxing heteronuclei. Therefore, even during measurements, an NMR spectrometer stands still for most of the time, simply waiting for the system to relax. This problem can be partly alleviated by the “moving tube” NMR1 or the addition of a paramagnetic relaxation agent.2–5 However, by the latter approach transverse relaxation is increased too, leading to broader signals. For NH-detected experiments on proteins the use of band-selective pulses also leads to enhanced R1 relaxation.6–8 However, all these approaches aimed at reducing the time between acquisition and start of the next excitation still require an interscan delay the duration of which is often several times the length of the actual NMR experiment. Theoretically, saturation for short relaxation delays can also be reduced by using smaller pulse-angles and purge gradients,7,9,10 however different relaxation rates for different resonances in one molecule will also lead to varying signal intensities.
Typical mechanistic tools to identify and quantify the components occurring during fast transient reaction by NMR are rapid-injection11–14 and stopped flow techniques.15,16 Thereby, high temporal resolution can be achieved by repeating the experiment several times and changing the time between mixing and data acquisition. However, this approach hinges on a very careful experimental setup to allow a completely reproducible repetition of the experiment and large sample amounts as each time point is obtained from a separate measurement. It becomes impractical when many time points need to be acquired.
Here we present a new method for the fast acquisition of 1D NMR spectra where no relaxation delay is needed at all (Fig. 1a). Irradiation is achieved by a selective pulse applied during a weak pulsed field gradient. The gradient leads to a spatially dependent frequency shift across the NMR tube (Fig. 1b).17,18 The overall induced frequency shift Δν (in Hertz) is given by Δν = γ × G × s, where s is the length of the sensitive volume (in centimeters), G is the strength of the gradient (gauss per centimeter) and γ is the gyromagnetic ratio (second−1 gauss−1) of the observed nucleus. Thereby it is possible to excite the whole spectrum by a single selective pulse as it irradiates different resonances in different slices of the sample. For excitation we typically use an EBURP-2 pulse19 with an excitation bandwidth of 300 Hz. A very similar scheme for “nonselective” excitation by selective pulses has been used previously for broadband homonuclear decoupling.18,20,21 An important feature of the continuous NMR approach is the shifting of the excitation frequency after each scan (Fig. 1b). Therefore, different signals are excited in a slice in the next scan, while previously excited signals can start to relax. The overall time per scan is reduced to the length of the selective pulse and the acquisition time. A further benefit of proteins is the faster T1 relaxation, if only a subset of spins is excited.6 In addition any magnetic field inhomogeneities along the z-direction, which lead to signal broadening in regular 1D spectra, are irrelevant because each signal is only excited in a small slice as previously pointed out by Freeman.22 Because of the shifting of the excitation frequency and to ensure that the whole spectrum is excited in each scan uniformly, the strength of the gradient has to be increased to G = (SW + NI × ΔSPOFFS)/γ × s, where SW is the sweep width, NI is the number of increments (scans) and ΔSPOFFS is the frequency shift between scans. The excitation frequency is changed NI times and then starts from its initial value. Thus, all spins can relax for a duration of NI times the complete experimental time per increment. Systems with high T1 values simply need more increments to allow more time for relaxation, achieved by stronger gradients and additional excitation frequencies. However, the stronger the gradient the smaller the excited slices are and in turn the lower the peak intensities. Assuming an excitation bandwidth of Δω (Hz), the sensitivity loss compared to a non-selectively excited spectrum can be estimated by (Δω/γ × G × s) × 100%. An experimental example is shown in Fig. S2 (ESI†). Nevertheless, the lower sensitivity can be overcome by recent progress in NMR hardware, like cryoprobes and stronger magnetic fields. As an example for the signal stability of the presented method Fig. S1 (ESI†) shows the first 20 spectra of a mixture of imidazole and alanine in phosphate buffer acquired with a sweep width of 8012.82 Hz and 512 complex data points, resulting in a digital resolution of 15.65 Hz per point and an acquisition time of 32 ms. In combination with a 300 Hz EBURP-2 (16.5 ms) pulse, this sums up to 48.5 ms per scan. Therefore, the twenty spectra in Fig. S1 (ESI†) correspond to an overall experimental time of only 970 ms. The uniform signal intensities confirm the complete absence of saturation. In contrast, saturation can be observed for slowly relaxing signals, when using small flip angle pulses23 (see Fig. S3, ESI†). The presented pulse sequence opens up the way for the continuous acquisition of quantitative, relaxation independent, 1D spectra with a high repetition rate, rendering it a convenient tool for kinetic studies. Sample volume changes caused by the rapid injection technique lead to a decrease in magnetic field homogeneity. One particularly important feature of the slice selective excitation is the above-mentioned independence of poor shimming in the z-direction. In contrast to regular NMR spectra (e.g. using small flip angle pulses) spatially selective spectra are significantly less affected by line broadening due to sample volume changes (see Fig. S4, ESI†).
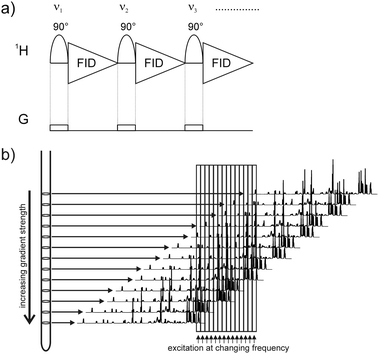 | ||
| Fig. 1 The pulse sequence of the continuous NMR experiment is shown in (a) for the first three increments. The frequency of the selective pulse is shifted between consecutive increments. A weak gradient field is applied during the excitation, which results in a spatially selective shift of the whole spectrum as indicated in (b). Selective excitation therefore leads to the excitation of the whole spectrum but each signal is excited in a different slice of the NMR tube. | ||
NMR spectroscopy has often been used for kinetic investigations of unstable reaction intermediates and mechanisms in organic chemistry12,23 but has also been extended to the field of biochemistry, where it has been an important tool for the investigation of for example enzyme reactions or protein folding.15,24 As a biochemical application of the presented method we have used it to study fast kinetics of pH-induced protein unfolding in myoglobin using a rapid-injection device (see Fig. S5, ESI†) built similar to the model described by Hore et al.14
Myoglobin, which is a model system for protein folding, consists of eight helices, with an iron-chelating, non-covalently attached heme group in its center.25 Both folding and unfolding of apo- as well as of holo-myoglobin from different organisms have been extensively studied using techniques like NMR and mass spectrometry.26,27 The unfolding of horse heart myoglobin (hMg) under acidic conditions was reported to involve a two step unfolding process.26 After starting the continuous acquisition experiment acetic acid was injected into a 5 mm Shigemi tube containing the hMg solution buffered at pH 6.5 right inside the NMR magnet. Complete mixing with the used rapid injection equipment is achieved within a dead time of ∼100 ms as described by Mok et al.14 The acquisition of 1024 data points, a sweep width of 8417.51 Hz and excitation with a 300 Hz EBURP-2 pulse sum up to a total time per increment of 78 ms. The frequency was shifted by 500 Hz between increments to make sure no signals are excited again in two consecutive increments, thereby allowing 16 increments per cycle. The total acquisition time of one cycle of 1.2 seconds is enough for hMg to prevent any significant intensity changes between the individual cycles as shown in Fig. S6 (ESI†). The time-course during acid-induced unfolding of hMg can be seen in Fig. 2.
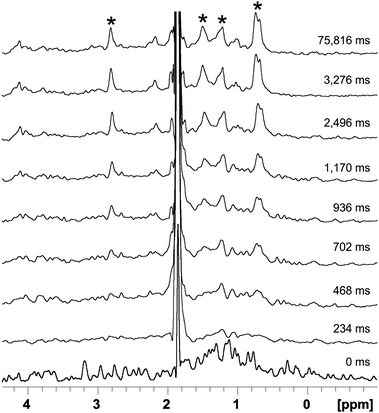 | ||
| Fig. 2 Time course of the acid induced unfolding reaction of myoglobin. The molten globule state indicated by broad signals forms within the dead time of the experiment (∼100 ms) and can be seen within the first few 100 ms. Roughly 400 ms after the injection, signals of the fully unfolded state start to appear and the protein is fully unfolded after a few seconds. Data were acquired every 78 ms. Only a few selected spectra are shown. Strong signals of the unfolded conformation which are broadened extensively in the molten globular state are indicated by asterisks. | ||
In the high-field region of the 1H proton spectrum a series of well-resolved signals are found down to −1.0 ppm, typical for a well-structured protein. The structure of the protein collapses within the mixing dead time (>100 ms). Right after the initial collapse of the structure the protein shows very broad and low intensity peaks. This is typical for a molten globule state, where the broad peaks are believed to be a result of conformational fluctuations on a millisecond time scale throughout the molten globule. These NMR characteristics of the molten globule state have been previously described for α-lactalbumins.28,29 Roughly 400 ms after the injection signals of the fully unfolded state start to appear. Our observations are in good agreement with previous studies where an unfolding intermediate of myoglobin has been reported. It has been hypothesized that parts of the secondary structure are stabilized by the heme group leading to a molten globule state in the beginning of the unfolding.26 Afterwards the random coil structure prevails with its typical intense collective methyl peak at 0.9 ppm. As more and more molecules completely unfold the methyl peak's intensity becomes larger and finally reaches its maximum intensity after 3 seconds.
In principle, the presented technique could also be extended to heteronuclear multi-dimensional NMR, where several dozens of increments could be acquired in one second. Such an experiment could compete, if sensitivity is not limiting, with currently employed concepts for fast multidimensional experiments.7,30–32
In conclusion, we have shown that, by using a series of frequency-shifted selective pulses applied during a weak pulsed field gradient, it is possible to obtain quantitative 1D proton spectra without the need for any relaxation delay between consecutive scans. Thus, it is possible to achieve an extremely high temporal resolution in NMR spectroscopy, only limited by the time needed for the selective pulse and the data acquisition. However, the huge increase in data acquisition speed is traded against low sensitivity. This continuous acquisition NMR experiment should also prove valuable for smaller molecules where sensitivity is usually less demanding and 1D spectra contain a relatively high information density. Besides rapid-injection techniques this method would be very well suited for light-induced reactions with a much shorter reaction initiation dead-time.
Financial support to K.Z. from the Austrian Science Foundation (FWF) (DK Molecular Enzymology W901 and project number P24742) is gratefully acknowledged. We would like to thank Peter J. Hore and Charles Jones (Department of Chemistry, Oxford University) for advice and technical drawings of their rapid injection device.
Notes and references
- K. J. Donovan, M. Allen, R. W. Martin and A. J. Shaka, J. Magn. Reson., 2012, 219, 41–45 CrossRef CAS.
- D. J. Cookson and B. E. Smith, J. Magn. Reson., 1984, 57, 355–368 CAS.
- T. Madl, W. Bermel and K. Zangger, Angew. Chem., Int. Ed., 2009, 48, 8259–8262 CrossRef CAS.
- G. Pintacuda and G. Otting, J. Am. Chem. Soc., 2001, 124, 372–373 CrossRef.
- N. P. Wickramasinghe, M. Kotecha, A. Samoson, J. Past and Y. Ishii, J. Magn. Reson., 2007, 184, 350–356 CrossRef CAS.
- K. Pervushin, B. Vögeli and A. Eletsky, J. Am. Chem. Soc., 2002, 124, 12898–12902 CrossRef CAS.
- P. Schanda, V. Forge and B. Brutscher, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11257–11262 CrossRef CAS.
- P. Schanda, H. Van Melckebeke and B. Brutscher, J. Am. Chem. Soc., 2006, 128, 9042–9043 CrossRef CAS.
- R. R. Ernst, G. Bodenhausen and A. Wokaun, Principles of Nuclear Magnetic Resonance in One and Two Dimensions, Clarendon Press, Oxford, 1994 Search PubMed.
- A. Bax, T. Mehlkopf and J. Smidt, J. Magn. Reson., 1980, 41, 502–506 CAS.
- S. E. Denmark, B. M. Eklov, P. J. Yao and M. D. Eastgate, J. Am. Chem. Soc., 2009, 131, 11770–11787 CrossRef CAS.
- J. F. McGarrity and J. Prodolliet, J. Org. Chem., 1984, 49, 4465–4470 CrossRef CAS.
- J. F. McGarrity, J. Prodolliet and T. Smyth, Org. Magn. Reson., 1981, 17, 59–65 CrossRef CAS.
- K. H. Mok, T. Nagashima, I. J. Day, J. A. Jones, C. J. V. Jones, C. M. Dobson and P. J. Hore, J. Am. Chem. Soc., 2003, 125, 12484–12492 CrossRef CAS.
- P. J. Hore, S. L. Winder, C. H. Roberts and C. M. Dobson, J. Am. Chem. Soc., 1997, 119, 5049–5050 CrossRef CAS.
- R. O. Kuhne, T. Schaffhauser, A. Wokaun and R. R. Ernst, J. Magn. Reson., 1979, 35, 39–67 Search PubMed.
- R. Freeman, Concepts Magn. Reson., Part A, 2011, 38, 1–6 Search PubMed.
- K. Zangger and H. Sterk, J. Magn. Reson., 1997, 124, 486–489 CrossRef CAS.
- H. Geen and R. Freeman, J. Magn. Reson., 1991, 93, 93–141 Search PubMed.
- J. A. Aguilar, S. Faulkner, M. Nilsson and G. A. Morris, Angew. Chem., Int. Ed., 2010, 49, 3901–3903 CrossRef CAS.
- M. Nilsson and G. A. Morris, Chem. Commun., 2007, 933–935 RSC.
- A. Bax and R. Freeman, J. Magn. Reson., 1980, 37, 177–181 CAS.
- S. E. Denmark, B. J. Williams, B. M. Eklov, S. M. Pham and G. L. Beutner, J. Org. Chem., 2010, 75, 5558–5572 CrossRef CAS.
- C. M. Dobson and P. J. Hore, Nat. Struct. Biol., 1998, 5, 504–507 CrossRef CAS.
- S. V. Evans and G. D. Brayer, J. Mol. Biol., 1990, 213, 885–897 CrossRef CAS.
- O. O. Sogbein, D. A. Simmons and L. Konermann, J. Am. Soc. Mass Spectrom., 2000, 11, 312–319 CrossRef CAS.
- T. Uzawa, C. Nishimura, S. Akiyama, K. Ishimori, S. Takahashi, H. J. Dyson and P. E. Wright, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13859–13864 CrossRef CAS.
- J. Baum, C. M. Dobson, P. A. Evans and C. Hanley, Biochemistry, 1989, 28, 7–13 CrossRef CAS.
- H. I. Rösner and C. Redfield, J. Mol. Biol., 2009, 394, 351–362 CrossRef.
- L. Frydman, T. Scherf and A. Lupulescu, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15858–15862 CrossRef CAS.
- E. Kupce and R. Freeman, J. Biomol. NMR, 2003, 25, 349–354 CrossRef CAS.
- P. Schanda and B. Brutscher, J. Am. Chem. Soc., 2005, 127, 8014–8015 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and continuous NMR spectra. See DOI: 10.1039/c3cc39107h |
| This journal is © The Royal Society of Chemistry 2013 |