 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSulfur doped Co/SiO2catalysts for chirally selective synthesis of single walled carbon nanotubes†
Hong
Wang
,
Kunli
Goh
,
Rui
Xue
,
Dingshan
Yu
,
Wenchao
Jiang
,
Raymond
Lau
and
Yuan
Chen
*
School of Chemical and Biomedical Engineering, Nanyang Technological University, Singapore 637459, Singapore. E-mail: chenyuan@ntu.edu.sg; Tel: +65-63168939
First published on 21st January 2013
Abstract
Monochiral single-walled carbon nanotubes have many potential uses, especially in the field of electronics and medical applications. Here we demonstrate a method to activate three types of Co/SiO2catalystsvia sulfur-doping for the synthesis of tubes with a narrow chiral distribution around (9,8) chirality.
Electronic and optical properties of single-walled carbon nanotubes (SWCNTs) are correlated with their geometric structures, i.e. chirality index (n,m).1 Thus, many potential applications require the use of monochiral SWCNTs. Chirally selective synthesis of nanotubes is the ultimate goal in SWCNT production, which remains a great challenge. In common growth methods, i.e.chemical vapor deposition (CVD), carbon precursors decompose at high temperature to form atomic carbon species, which then nucleate into one-dimensional tubes on catalytic particles. Theoretical studies suggest that catalysts play a critical role in the (n,m) selectivity.2–7 To control SWCNT (n,m) structures, the size of catalytic particles need to be first confined in a suitable range.7–9 However, metal species are mobile and form particles of various sizes at high growth temperatures, which eventually results in the growth of multiwalled carbon nanotubes (MWCNTs), graphite and amorphous carbon.
Co catalysts supported on silica substrates (Co/SiO2) are the most often used catalysts for (n,m) selective growth of SWCNTs.10–20 Several approaches have been used to control the nucleation of Co, e.g. stabilizing Co clusters in bimetallic alloys,10,12,20 incorporating Co ions into mesoporous substrates,11,14,16 forming dispersed surface Co silicates,17 anchoring reduced Co atoms by unreduced Co ions,18 and creating strong interfacial Co–Si interactions.19 Most of these approaches only result in the selective growth of small-diameter (6,5) tubes. Some previous studies had shown that when sulfur (S) species are added to carbon precursors, it can promote the growth rate and yield of the nanotubes, and change MWCNT diameters.21–24 However, to the best of our knowledge, no work has been reported on the direct doping of S in solid catalysts to control metal particles for SWCNT growth.
In this work, we have demonstrated that non-selective Co/SiO2catalysts can be converted into efficient chiral selective catalysts by S doping. SWCNTs were characterized by photoluminescence (PL), UV-vis-near-infrared (UV-vis-NIR) absorption and Raman spectroscopies. Catalysts were characterized by elemental analysis, H2 temperature programmed reduction (H2-TPR), and UV-vis diffuse reflectance spectroscopy. The molecular structural changes of Co species on SiO2 caused by S doping are believed to be responsible for the chiral selectivity.
Three Co/SiO2catalysts with 1 wt% Co were prepared by the impregnation method using Co precursors, including Co(II) acetylacetonate (Co(acac)2, Sigma-Aldrich, 97%), Co(II) chloride (CoCl2, Alfa Aesar, 97%), and Co(II) nitrate hexahydrate (Co(NO3)2·6H2O, Sigma-Aldrich, 99.999%). They are denoted as CoACAC/SiO2, CoCl/SiO2, and CoN/SiO2, respectively. To add S, the calcined Co/SiO2catalysts were impregnated by dilute sulfuric acid (H2SO4, 0.04 mol L−1) at 8 mL solution per g catalyst ratio for 1 h. The resulting S doped catalysts were marked as CoACAC/SiO2/S, CoCl/SiO2/S, and CoN/SiO2/S, respectively (see ESI† for details). Catalysts were introduced into a CVD reactor, and first reduced under pure H2 (1 bar, 50 sccm) from room temperature to 540 °C at 20 °C min−1, and then further heated to 780 °C under an Ar flow (1 bar, 50 sccm). At 780 °C, pressured CO (6 bar, 200 sccm) replaced Ar, and the SWCNT growth lasted for 1 h. As-synthesized SWCNTs with catalysts were first characterized by Raman spectroscopy, and then dissolved in NaOH aqueous solution (1.5 mol L−1) to remove SiO2. The carbon deposits were then dispersed in 2 wt% sodium dodecyl benzene sulfate (SDBS, Aldrich) D2O solution. The SWCNT suspension obtained after centrifugation at 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g was characterized by PL and UV-vis-NIR absorption spectroscopies (see ESI† for details).
000 g was characterized by PL and UV-vis-NIR absorption spectroscopies (see ESI† for details).
PL maps in Fig. 1a–f show that two undoped Co/SiO2catalysts (CoACAC/SiO2 and CoCl/SiO2) resulted in small-diameter tubes (<0.9 nm), such as (6,5), (7,5), (7,6) and (8,4). CoN/SiO2 is not active for SWCNT growth. This is in agreement with previous studies using various SiO2 supported Co catalysts.15,17,25 In contrast, after doping with S, the major (n,m) products are large-diameter tubes (>1.1 nm), such as (9,8), (9,7), (10,6), and (10,9). The abundance of these four species calculated using their PL intensity is 52.4–69.1% of all semiconducting species identified, out of which, 32.7–40.5% is (9,8) (see Tables S1–S3 in the ESI†). PL results were corroborated by UV-vis-NIR absorption spectra. Fig. 1g shows that SWCNTs from CoACAC/SiO2 and CoCl/SiO2 have intense absorption peaks at 992, 1025 and 1137 nm from (6,5), (7,5), (7,6), and (8,4). SWCNTs grown on CoN/SiO2 have weak absorption peaks. In contrast, Fig. 1h shows that SWCNTs grown on S doped catalysts all have strong absorption peaks at 1420 and 818 nm, corresponding to the ES11 and ES22 transitions of (9,8). A few other absorption peaks below 700 nm can be assigned to either the EM11 transition of metallic (9,6) (1.02 nm) and (10,10) (1.36 nm) or the ES22 transition of semiconducting (6,5). Since the intensity of (6,5) PL peaks in Fig. 1d–f is low, the absorption peaks below 700 nm in Fig. 1h are likely to be from those metallic tubes. SWCNTs were further characterized by Raman spectroscopy under two excitation lasers (785 and 514 nm). Raman results (see Fig. S1 in the ESI†) agree with the findings from PL and UV-vis-NIR. Overall, the three spectroscopic techniques have demonstrated that S doping can shift the (n,m) selectivity of Co/SiO2catalysts from small-diameter tubes near (6,5) to large-diameter tubes with a narrow distribution around (9,8).
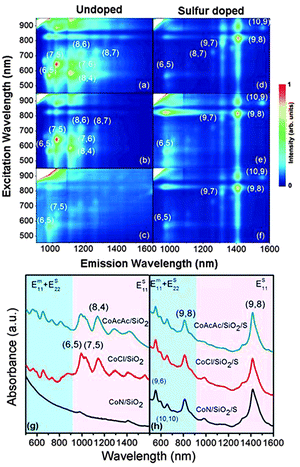 | ||
| Fig. 1 PL maps and UV-vis-NIR absorption spectra of SDBS-dispersed SWCNTs grown on undoped and S doped Co/SiO2catalysts. PL maps: (a) CoACAC/SiO2, (b) CoCl/SiO2, (c) CoN/SiO2, (d) CoACAC/SiO2/S, (e) CoCl/SiO2/S, and (f) CoN/SiO2/S. Some major (n,m) species identified on PL maps are marked. Absorption spectra: undoped (g) and S doped (h). The shaded pink indicates the ES11 absorption band and the shaded blue shows the overlapping ES22 and EM11 bands. | ||
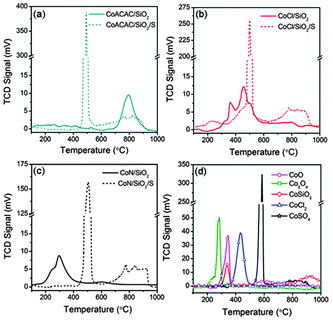 | ||
| Fig. 2 H2-TPR profiles of undoped and S doped Co/SiO2catalysts and several Co references (CoO, Co3O4, CoSiO3, CoCl2 and CoSO4·7H2O). | ||
Co species deposited on SiO2 would first be partially reduced in H2, and then nucleated into Co nanoparticles to initiate SWCNT growth. The change in (n,m) selectivity shown in Fig. 1 may be attributed to the changes in Co species caused by S doping. Firstly, we conducted an elemental analysis of S doped catalysts. The S content in CoACAC/SiO2/S, CoCl/SiO2/S, and CoN/SiO2/S was found to be 0.91, 1.17 and 0.83 wt%, respectively. This confirms the existence of S. Next, H2-TPR was employed to study the reducibility of Co species. Fig. 2 shows that CoACAC/SiO2 displays a peak around 797 °C, which is due to the surface Co silicate.17 CoCl/SiO2 has multiple peaks at 360–800 °C, which may come from the reduction of CoO, CoCl2, and surface Co silicate. CoN/SiO2 possesses a broad peak around 290 °C which can be attributed to Co3O4 and CoO. In contrast, all the TPR profiles of the three S doped Co/SiO2catalysts have a sharp peak at 493–506 °C, similar to our recently reported chiral selective CoSO4/SiO2catalyst.26 In addition, it is observed that the CoOx peaks of undoped catalysts become significantly smaller after S doping, and new peaks around 800 °C appear on CoCl/SiO2/S and CoN/SiO2/S. This observation suggests the formation Co hydrosilicate or surface Co silicate,27 while the 797 °C peak of CoACAC/SiO2 becomes smaller. Lastly, UV-vis diffuse reflectance spectroscopy was used to probe the surface chemistry of the catalysts. As shown in Fig. S2 in the ESI,† CoACAC/SiO2 has two peaks around 570 and 650 nm, suggesting the formation of surface Co silicates.28 The spectrum of CoCl/SiO2 shows two broad peaks at 550 and 720 nm, indicating the presence of CoOx and CoCl2. The spectrum of CoN/SiO2 is similar to that of Co3O4, having two broad peaks at ∼400 and 720 nm, which can be assigned to the transitions of octahedral configured Co3+ ions.29 In contrast, all the three S doped Co/SiO2catalysts have a broad peak around 535 nm, similar to that of CoSO4, and this suggests the existence of Co species bonded to SO42−.
Doping sulfate ions into metal oxides has created various solid acid catalysts, such as SO42−/ZrO2, SO42−/TiO2, and SO42−/Fe2O3.30,31 Based on our characterization of the Co/SiO2catalysts, we propose the following mechanism to explain their (n,m) selectivity in SWCNT growth. As shown in Fig. 3, undoped Co/SiO2catalysts contain CoOx, Co hydrosilicate, and surface Co silicates, which are evident from their H2-TPR profiles and UV-visspectra. Surface Co silicates on CoACAC/SiO2 and CoCl/SiO2 would be reduced and nucleated into small Co nanoparticles, which are selective toward small-diameter SWCNTs,17 as shown in Fig. 1. CoOx on CoN/SiO2 is reduced to form large Co particles, which are not selective to SWCNTs. Doping S through H2SO4 leads to the formation of chelating bidentate SO42−, where one S atom is linked to one Co atom through two O atoms, a common structure found in sulfate promoted metal oxidecatalysts.30 This is supported by the sharp peaks at 493–506 °C in the TPR profiles and the broad peak around 535 nm in the UV-vis spectra. Windle et al. showed that adding thiophene or carbon disulfide in gas phase would restrict the growth of Fe particles for selective growth of (9,9).32 Similarly, we propose that the co-existence of S atoms near Co atoms may limit the nucleation of Co and/or form Co–S compounds, which changes the selectivity of the catalysts to favor the formation of (9,8). As discussed in our previous study,26 the selectivity toward (9,8) may be attributed to the close match between carbon caps and the most stable Co particles in their size range, as well as the higher growth rate of high chiral angle tubes.33 As active Co nanoparticles are embedded under or near SiO2 surface, we are still unable to quantify their size and composition in transmission electron microscopy analysis (see Fig. S3 in the ESI†). Furthermore, we propose a reaction between H+ ions and CoOx, releasing Co ions to form well dispersed Co hydrosilicate and surface Co silicate on SiO2, which would increase the selectivity toward SWCNTs. This is suggested by the increased SWCNT selectivity of CoN/SiO2/S. To further verify the proposed mechanism, we doped CoN/SiO2 using (NH4)2SO4. We expected the same effect from SO42−, but selectivity toward SWCNTs may be compromised since NH4+ is less reactive than H+. As shown in Fig. S4–S7 in the ESI,† (NH4)2SO4 doped CoN/SiO2 can result in the growth of (9,8) nanotubes because of S doping. However, it is less selective to SWCNTs as compared to CoN/SiO2/S. This provides strong credibility to our proposed mechanism.
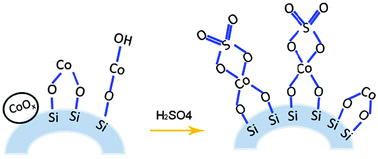 | ||
| Fig. 3 Schematic illustration of changes in Co species on Co/SiO2catalysts caused by S doping. | ||
In conclusion, we have demonstrated a method to convert three types of Co/SiO2catalysts, which are either inactive for the SWCNT growth or only selective to small-diameter nanotubes, into chirally selective catalysts to grow SWCNTs enriched with large-diameter (9,8) tubes (up to 40.5%) by doping catalysts with S. We also proposed in the mechanism that S atoms near Co atoms assist the formation of Co nanoparticles which are selective to (9,8) tubes. Moreover, H+ ions may react with CoOx to form well dispersed Co hydrosilicate and surface Co silicate on SiO2, which increases the selectivity to SWCNTs.
This work is supported by National Research Foundation (NRF-CRP2-2007-02) and Ministry of Education, Singapore (MOE2011-T2-2-062). We thank Prof. Qiang Zhang of Tsinghua University for the TEM analyses of our samples.
Notes and references
- R. H. Baughman, A. A. Zakhidov and W. A. de Heer, Science, 2002, 297, 787–792 CrossRef CAS.
- S. Irle, Y. Ohta, Y. Okamoto, A. J. Page, Y. Wang and K. Morokuma, Nano Res., 2009, 2, 755–767 CrossRef CAS.
- H. Dumlich and S. Reich, Phys. Rev. B, 2010, 82, 085421 CrossRef.
- E. C. Neyts, A. C. T. van Duin and A. Bogaerts, J. Am. Chem. Soc., 2011, 133, 17225–17231 CrossRef CAS.
- Q. Yuan, Z. Xu, B. I. Yakobson and F. Ding, Phys. Rev. Lett., 2012, 108, 245505 CrossRef.
- D.A. Gómez-Gualdrón, G. D. McKenzie, J. F. J. Alvarado and P. B. Balbuena, ACS Nano, 2012, 6, 720–735 CrossRef.
- M. F. C. Fiawoo, A. M. Bonnot, H. Amara, C. Bichara, J. Thibault-Pénisson and A. Loiseau, Phys. Rev. Lett., 2012, 108, 195503 CrossRef.
- C. L. Cheung, A. Kurtz, H. Park and C. M. Lieber, J. Phys. Chem. B, 2002, 106, 2429–2433 CrossRef CAS.
- A. G. Nasibulin, P. V. Pikhitsa, H. Jiang and E. I. Kauppinen, Carbon, 2005, 43, 2251–2257 CrossRef CAS.
- S. M. Bachilo, L. Balzano, J. E. Herrera, F. Pompeo, D. E. Resasco and R. B. Weisman, J. Am. Chem. Soc., 2003, 125, 11186–11187 CrossRef CAS.
- S. Lim, D. Ciuparu, Y. Chen, L. Pfefferle and G. L. Haller, J. Phys. Chem. B, 2004, 108, 20095–20101 CrossRef CAS.
- Y. Miyauchi, S. Chiashi, Y. Murakami, Y. Hayashida and S. Maruyama, Chem. Phys. Lett., 2004, 387, 198–203 CrossRef CAS.
- B. Wang, C. H. P. Poa, L. Wei, L. J. Li, Y. Yang and Y. Chen, J. Am. Chem. Soc., 2007, 129, 9014–9019 CrossRef CAS.
- Y. Chen, L. Wei, B. Wang, S. Y. Lim, D. Ciuparu, M. Zheng, J. Chen, C. Zoican, Y. H. Yang, G. L. Haller and L. D. Pfefferle, ACS Nano, 2007, 1, 327–336 CrossRef CAS.
- B. Wang, Y. Yang, L. J. Li and Y. Chen, J. Mater. Sci., 2009, 44, 3285–3295 CrossRef CAS.
- H. Wang, B. Wang, X. Quek, L. Wei, J. Zhao, L. Li, M. Chan-Park, Y. Yang and Y. Chen, J. Am. Chem. Soc., 2010, 132, 16747–16749 CrossRef CAS.
- N. Li, X. M. Wang, S. Derrouiche, G. L. Haller and L. D. Pfefferle, ACS Nano, 2010, 4, 1759–1767 CrossRef CAS.
- M. S. A. He, A. I. Chernov, P. V. Fedotov, E. D. Obraztsova, E. Rikkinen, Z. Zhu, J. Sainio, H. Jiang, A. G. Nasibulin, E. I. Kauppinen, M. Niemela and A. O. I. Krause, Chem. Commun., 2011, 47, 1219–1221 RSC.
- M. Fouquet, B. C. Bayer, S. Esconjauregui, R. Blume, J. H. Warner, S. Hofmann, R. Schlogl, C. Thomsen and J. Robertson, Phys. Rev. B, 2012, 85, 235411 CrossRef.
- B. Liu, W. Ren, S. Li, C. Liu and H. M. Cheng, Chem. Commun., 2012, 48, 2409–2411 RSC.
- L. Ci, J. Wei, B. Wei, J. Liang, C. Xu and D. Wu, Carbon, 2001, 39, 329–335 CrossRef CAS.
- W. Ren, F. Li, S. Bai and H. M. Cheng, J. Nanosci. Nanotechnol., 2006, 6, 1339–1345 CrossRef CAS.
- J. Wei, H. Zhu, Y. Jia, Q. Shu, C. Li, K. Wang, B. Wei, Y. Zhu, Z. Wang and J. Luo, Carbon, 2007, 45, 2152–2158 CrossRef CAS.
- B. Yu, C. Liu, P. X. Hou, Y. Tian, S. Li, B. Liu, F. Li, E. I. Kauppinen and H. M. Cheng, J. Am. Chem. Soc., 2011, 133, 5232–5235 CrossRef CAS.
- W. E. Alvarez, B. Kitiyanan, A. Borgna and D. E. Resasco, Carbon, 2001, 39, 547–558 CrossRef CAS.
- H. Wang, L. Wei, F. Ren, Q. Wang, L. D. Pfefferle, G. L. Haller and Y. Chen, ACS Nano, 2013, 7, 614–626 CrossRef CAS.
- E. v. Steen, G. S. Sewell, R. A. Makhothe, C. Micklethwaite, H. Manstein, M. de Lange and C. T. O'Connor, J. Catal., 1996, 162, 220–229 CrossRef.
- A. Khodakov, J. Girardon, A. Griboval-Constant, A. Lermontov and P. Chernavskii, Stud. Surf. Sci. Catal., 2004, 147, 295–300 CrossRef CAS.
- Y. Brik, M. Kacimi, M. Ziyad and F. Bozon-Verduraz, J. Catal., 2001, 202, 118–128 CrossRef CAS.
- T. Yamaguchi, Appl. Catal., 1990, 61, 1–25 CrossRef CAS.
- T. Yamaguchi, T. Jin, T. Ishida and K. Tanabe, Mater. Chem. Phys., 1987, 17, 3–19 CrossRef CAS.
- R. M. Sundaram, K. K. K. Koziol and A. H. Windle, Adv. Mater., 2011, 23, 5064–5068 CrossRef CAS.
- F. Ding, A. R. Harutyunyan and B. I. Yakobson, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2506–2509 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cc38973a |
| This journal is © The Royal Society of Chemistry 2013 |