 Open Access Article
Open Access ArticleEnantioselective total synthesis of virosaine A and bubbialidine†‡
Hideki Miyatake-Ondozabal, Linda M. Bannwart and Karl Gademann*
Department of Chemistry, University of Basel, St. Johanns-Ring 19, Basel 4056, Switzerland. E-mail: karl.gademann@unibas.ch; Tel: +41 612671144
First published on 18th January 2013
Abstract
The first enantioselective total syntheses of virosaine A and bubbialidine are described. Key transformations include the formation of a tetracyclic intermediate via an intramolecular aza-Michael addition, generation of a N-hydroxy-pyrrolidine through a Cope elimination and an intramolecular [1,3]-dipolar cycloaddition to generate a complex 7-oxa-1-azabicyclo[3.2.1]octane ring system.
The securinega alkaloids are a family of bridged tetracyclic natural products occurring in the plants of the Securinega, Phyllanthus, Flueggea and other genera in the Euphorbiaceae family.1 Recently, two new birdcage-shaped alkaloids with unprecedented skeletal structures were isolated, namely virosaine A (1) and virosaine B (2), from the twigs and leaves of Flueggea virosa (Fig. 1).2 The unique structural features of these pseudoenantiomers are characterized by their densely functionalized, stereochemically complex architecture featuring an unusual tetracyclic core incorporating a trihydro-1,2-oxazine ring. Neither 1 nor 2 showed cytotoxic activity against selected cancer cell lines (MCF-7, MDA-MB-231, HepG2, HepG2/ADM, HL-60, K562 and Hep2).2
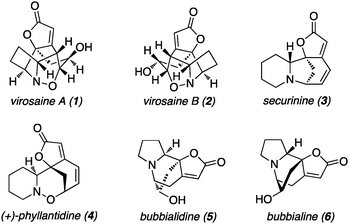 | ||
| Fig. 1 Virosaines A, B and examples of related alkaloids. | ||
Among this family of natural products, securinine (3) is the most abundant and widely spread alkaloid possessing an impressive range of biological activity including neurotransmitter gamma-aminobutyric acid (GABA) receptor antagonism,3in vivo CNS activity and anti-malarial and anti-bacterial activities.4,5 Due to its remarkable biological activities and intriguing molecular structure, numerous total syntheses have been reported to date.1b,6 Conversely, a related yet much rarer securinega alkaloid (+)-phyllantidine (4) has a similar cyclic hydroxylamine scaffold to virosaines A (1) and B (2) and only one total synthesis has been published due to its complex architecture.7 During the preparation of this manuscript, the first total synthesis of virosaine B was reported by Yang, Li and coworkers.8 Two other putatively related alkaloids bubbialidine (5) and bubbialine (6) were isolated from the leaves of Zygogynum pauciflorum by Potier et al. in 1990.9 There is no reported publication for the synthesis of virosaine A and bubbialidine to date. In this communication, we report the first total syntheses of virosaine A (1) and bubbialidine (5).
Our brief retrosynthetic analysis is illustrated in Scheme 1. The main synthetic strategies are the vinylogous Mannich reaction, an intramolecular aza-Michael addition, a late-stage regioselective oxidation of the pyrrolidine moiety to the nitrone and the subsequent intramolecular [1,3]-dipolar cycloaddition. Inspired by the proposed biosynthesis of virosaines suggested by Zhang, Ye and coworkers,2 we envisaged that nitrone 7 should undergo a stereoselective intramolecular cycloaddition to form the complex tetracyclic core 1, creating three new stereogenic centres. A regioselective oxidation could be achieved in N-hydroxy-pyrrolidine 8 leading to 7. Following a synthetic strategy described by Magnus et al.,10 oxidation precursor 8 should be available from tetracycle 9 through an N-oxidation–Cope elimination sequence. An intramolecular aza-Michael addition of pyrrolidinyl-furanone 10 would allow the formation of 9, which will serve as a masked alkene intermediate enabling N-oxidation in the next step. Finally, a vinylogous Mannich reaction between aminol 11 and furanone 12 should provide the key intermediate 10 after t-butyloxycarbonyl (Boc) deprotection.
 | ||
| Scheme 1 Retrosynthesis of virosaine A and bubbialidine. | ||
The preparation of silyl-protected aquilegiolide (+)-12 was carried out following three reported publications (Schemes 2 and 3).11 The synthesis started with commercially available 1,4-cyclohexadiene 13. A three step procedure involving mono-epoxidation, ring-opening with cyanomethyllithium and acetylation gave the racemic acetate (±)-14 in 22% yield. Enzymatic kinetic resolution was then employed to generate enantiomerically enriched alcohol (−)-15 and acetate (+)-14 with 94.6% ee and 96.0% ee, respectively.11a
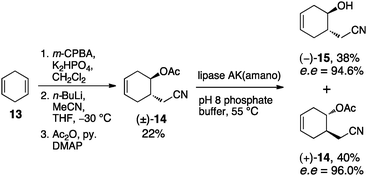 | ||
| Scheme 2 Enantioselective synthesis of (−)-15 and (+)-14. | ||
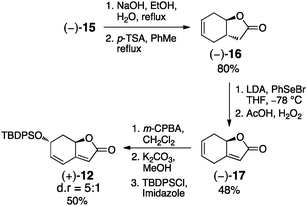 | ||
| Scheme 3 Synthesis of butenolide 12. | ||
Treatment of alcohol (−)-15 under basic conditions triggered the hydrolysis of the nitrile functionality (Scheme 3). Subsequent acid catalysed lactonisation with p-toluenesulfonic acid gave lactone (−)-16 in 80% yield over two steps. Following phenylselenation and oxidative elimination, butenolide (−)-17 was accessed in moderate yield.11b The silyl protected aquilegiolide (+)-12 was obtained by diastereoselective epoxidation (dr = 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), base-induced epoxide opening and silyl protection in good yields over three steps.11c
:1), base-induced epoxide opening and silyl protection in good yields over three steps.11c
The enantioselective synthesis of virosaine A (1) is described in Scheme 4. The first key transformation, a vinylogous Mannich reaction,12 between (+)-12 and aminol 1113 was achieved using triisopropylsilyl triflate as a Lewis acid, an elegant methodology reported by Busqué and coworkers.11c This resulted in the formation of solely two diastereoisomers (among the four possible) in 1:1 ratio in a yield of 90%. Pleasingly, the two isomers were separable by column chromatography allowing clean isolation of the desired adduct (−)-18. The Boc group was smoothly removed using a hydrogen chloride solution to give (−)-19 in a quantitative yield. To our surprise, the treatment of HCl salt (−)-19 with potassium hydrogenphosphate at elevated temperature facilitated an intramolecular aza-Michael addition14 to furnish the tetracycle (−)-9 in a remarkable yield of 90%.
 | ||
| Scheme 4 Enantioselective synthesis of virosaine A (1). | ||
This transformation enabled efficient formation of N-oxide 20 (supported by 1H-NMR characterization) in the next step using m-chloroperbenzoic acid and the alkene functionality was revealed under slightly acidic conditions to yield N-hydroxy-pyrrolidine (−)-8 in 77% yield over two steps.15 The next step was the construction of the nitrone unit 7 utilizing a convenient and mild method developed by Mukaiyama and coworkers.16
Gratifyingly, the use of N-t-butylbenzenesulfinimidoyl chloride 2117 at −78 °C resulted in a complete regioselective formation of nitrone 7 due to steric encumbrance and an immediate intramolecular [1,3]-dipolar cycloaddition15a,18,19 was observed. Finally, the removal of silyl group was performed using tetrabutylammonium fluoride to give virosaine A (1) in a good yield of 81% over two steps. The synthetic virosaine A (1) displayed identical physical and spectroscopic data to those reported in the literature.2 In addition, the first synthesis of bubbialidine (5) was also accomplished by silyl deprotection of tetracycle (−)-9 to give the target natural product in 92% yield (Scheme 5). The synthetic sample displayed identical physical and spectroscopic data to the reported values.9
 | ||
| Scheme 5 Enantioselective synthesis of bubbialidine (5). | ||
In summary, we report the first enantioselective total syntheses of virosaine A (1) and bubbialidine (5). The synthesis of virosaine A was achieved in 18 steps whereas bubbialidine was synthesized in 15 steps starting from readily available material. Our synthetic strategy can be highlighted by the intramolecular aza-Michael addition for the construction of the tetracycle (−)-9, Cope-elimination for a late-stage oxidation of the pyrrolidine unit, and an intramolecular cycloaddition reaction to build the 7-oxa-1-azabicyclo[3.2.1]octane ring core. Further application of this approach towards related natural products is currently under investigation.
We thank the Latsis Foundation for support of this work (National Latsis Prize 2011 to K.G.).
Notes and references
- (a) V. Snieckus, in The Alkaloids, ed. R. H. F. Manske, Academic Press, New York, 1973, vol. 14, pp. 425–503 Search PubMed; (b) S. M. Weinreb, Nat. Prod. Rep., 2009, 26, 758 RSC; (c) J. A. Beutler and A. N. Brubaker, Drugs Future, 1987, 12, 957–976 Search PubMed.
- B.-X. Zhao, Y. Wang, D.-M. Zhang, X.-J. Huang, L.-L. Bai, Y. Yan, J.-M. Chen, T.-B. Lu, Y.-T. Wang, Q.-W. Zhang and W.-C. Ye, Org. Lett., 2012, 14, 3096 CrossRef CAS.
- D. Rognan, T. Boulanger, R. Hoffmann, D. P. Vercauteren, J.-M. Andre, F. Durant and C.-G. Wermuth, J. Med. Chem., 1992, 35, 1969 CrossRef CAS.
- (a) E. Galvez-Ruano, M. H. Aprison, D. H. Robertson and K. B. Lapkowitz, J. Neurosci. Res., 1995, 42, 666 CrossRef CAS; (b) H. Weenen, M. H. H. Nkunya, D. H. Bray, L. B. Mwasumbi, L. S. Kinabo, V. A. Kilimali and J. B. Wijnberg, Planta Med., 1990, 56, 371 CrossRef CAS; (c) J. L. Mensah, I. Lagarde, C. Ceschin, G. Michel, J. Gleye and I. Fouraste, J. Ethnopharmacol., 1990, 28, 129 CrossRef CAS; (d) H. Tatematsu, M. Mori, T.-H. Yang, J.-J. Chang, T. T.-Y. Lee and K.-H. Lee, J. Pharm. Sci., 1991, 80, 325 CrossRef CAS.
- For an early review of the biological activities of these alkaloids, see: J. A. Beutler and A. N. Brubaker, Drugs Future, 1987, 12, 957 Search PubMed.
- (a) S. Saito, H. Yoshikawa, Y. Sato, H. Nakai, N. Sugimoto, Z. Horii, M. Hanaoka and Y. Tamura, Chem. Pharm. Bull., 1966, 14, 313 CrossRef CAS; (b) T. Honda, H. Namiki, M. Kudoh, N. Watanabe, H. Nagase and H. Mizutani, Tetrahedron Lett., 2000, 41, 5927 CrossRef CAS; (c) S. Liras, J. E. Davoren and J. Bordner, Org. Lett., 2001, 3, 703 CrossRef CAS; (d) B. Dhudshia, B. F. T. Cooper, C. L. B. Macdonald and A. N. Thadani, Chem. Commun., 2009, 463 RSC.
- C. A. Carson and M. A. Kerr, Angew. Chem., Int. Ed., 2006, 45, 6560 CrossRef CAS.
- H. Wei, C. Qiao, G. Liu, Z. Yang and C.-C. Li, Angew. Chem., Int. Ed., 2013, 52, 620 CrossRef CAS.
- A. Ahond, J. Guilhem, J. Hamon, J. Hurtado, C. Poupat, J. Pusset, M. Pusset, T. Sévenet and P. Potier, J. Nat. Prod., 1990, 53, 875 CrossRef CAS . For the isolation of related natural products, see: (a) P. J. Houghton, T. Z. Woldemariam, S. O'Shea and S. P. Thyagarajan, Phytochemistry, 1996, 43, 715 CrossRef CAS; (b) J. R. Patela, P. Tripathib, V. Sharmaa, N. S. Chauhana and V. K. Dixit, J. Ethnopharmacol., 2011, 138, 286 CrossRef.
- P. Magnus, J. Ródriguez-López, K. Mulholland and I. Matthews, Tetrahedron, 1993, 49, 8059 CrossRef CAS.
- (a) N. Kato, M. Inada, H. Sato, S. Ito, M. Shoji and M. Ueda, Tetrahedron Lett., 2007, 48, 7702 CrossRef CAS; (b) G. Audran and K. Mori, Eur. J. Org. Chem., 1998, 57 CrossRef CAS; (c) G. G. Bardaji, M. Canto, R. Alibes, P. Bayon, F. Busqué, P. De March, M. Figueredo and J. Font, J. Org. Chem., 2008, 73, 7657 CrossRef CAS.
- For applications of vinylogous Mannich reactions to alkaloid synthesis, see: (a) S. Martin and A. Liras, J. Am. Chem. Soc., 1993, 115, 10450 CrossRef CAS; (b) S. F. Martin, C. W. Clark and J. W. Corbett, J. Org. Chem., 1995, 60, 3236 CrossRef CAS; (c) S. F. Martin, C. W. Clark, M. Ito and M. Mortimore, J. Am. Chem. Soc., 1996, 118, 9804 CrossRef CAS; (d) S. F. Martin and S. K. Bur, Tetrahedron, 1999, 55, 8905 CrossRef CAS; (e) S. F. Martin, K. J. Barr, D. W. Smith and S. K. Bur, J. Am. Chem. Soc., 1999, 121, 6990 CrossRef CAS . For a review of the Mannich reaction, see: ; (f) M. Arend, B. Westermann and N. Risch, Angew. Chem., Int. Ed., 1998, 37, 1045 CrossRef CAS; (g) S. K. Bur and S. F. Martin, Tetrahedron, 2001, 57, 3221 CrossRef CAS.
- S. Peixoto, T. M. Nguyen, D. Crich, B. Delpech and C. Marazano, Org. Lett., 2010, 12, 4760 CrossRef CAS.
- For recent reviews in this area, see: (a) D. Enders, C. Wang and J. X. Liebich, Chem.–Eur. J., 2009, 15, 11058 CrossRef CAS; (b) P. R. Krishna, A. Sreeshailam and R. Srinivas, Tetrahedron, 2009, 65, 9657 CrossRef CAS; (c) J. Wang, P. Li, P. Y. Choy, A. S. C. Chan and F. Y. Kwong, ChemCatChem, 2012, 4, 917 CrossRef CAS.
- For examples of synthesis of N-hydroxy-pyrrolidines through Cope elimination, see: (a) J. J. Tufariello, Acc. Chem. Res., 1979, 12, 396 CrossRef CAS; (b) I. A. O'Neil, E. Cleator and D. J. Tapolczay, Tetrahedron Lett., 2001, 42, 8247 CrossRef CAS; (c) I. A. O'Neil, E. Cleator, V. E. Ramos, A. P. Chorlton and D. J. Tapolczay, Tetrahedron Lett., 2004, 45, 3655 CrossRef CAS; (d) G. L. Ellis, I. A. O'Neil, V. E. Ramos, S. B. Kalindjian, A. P. Chorlton and D. J. Tapolczay, Tetrahedron Lett., 2007, 48, 1687 CrossRef CAS.
- J. Matsuo, T. Shibata, H. Kitagawa and T. Mukaiyama, ARKIVOC, 2001, 58 CAS , part (x).
- For other application of this reagent in organic synthesis, see: (a) T. Mukaiyama, J. Matsuo and M. Yanagisawa, Chem. Lett., 2000, 1072 CrossRef CAS; (b) T. Mukaiyama, A. Kawana, Y. Fukuda and J. Matsuo, Chem. Lett., 2001, 390 CrossRef CAS; (c) J. Matsuo, D. Iida, K. Tatani and T. Mukaiyama, Bull. Chem. Soc. Jpn., 2002, 75, 223 CrossRef CAS; (d) N. Z. Burns, I. Krylova, R. N. Hannoush and P. S. Baran, J. Am. Chem. Soc., 2009, 131, 9172 CrossRef CAS.
- (a) R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 565 CrossRef; (b) R. Huisgen, J. Org. Chem., 1968, 33, 2291 CrossRef; (c) D. St. C. Black, R. F. Crozier and V. C. Davis, Synthesis, 1975, 205 CrossRef CAS . For application of intramolecular 1,3-dipolar cycloaddition in organic synthesis, see the excellent reviews by: ; (d) A. Padwa, Angew. Chem., Int. Ed. Engl., 1976, 15, 123 CrossRef; (e) A. Padwa and A. M. Schoffstall, Adv. Cycloaddit., 1990, 2, 1 CAS; (f) V. Naira and T. D. Suja, Tetrahedron, 2007, 63, 12247 CrossRef.
- For the preparation of functionalized isoxazolidines using Cope elimination–intramolecular nitrone cycloaddition, see: I. A. O'Neil, V. E. Ramos, G. L. Ellis, E. Cleator, A. P. Chorlton, D. J. Tapolczay and S. B. Kalindjian, Tetrahedron Lett., 2004, 45, 3659 CrossRef CAS.
Footnotes |
| † Dedicated to Professor Dr Christoph Tamm on the occasion of his 90th birthday. |
| ‡ Electronic supplementary information (ESI) available: For experimental procedures and compound characterization of all new compounds. See DOI: 10.1039/c3cc38783f |
| This journal is © The Royal Society of Chemistry 2013 |