 Open Access Article
Open Access ArticleDouble parallel dynamic resolution through lipase-catalyzed asymmetric transformation†
Yan Zhang, Lei Hu and Olof Ramström*
Department of Chemistry KTH - Royal Institute of Technology Teknikringen 30, Stockholm, Sweden. E-mail: ramstrom@kth.se; Fax: +46 8 7912333
First published on 25th January 2013
Abstract
Dynamic systems based on double parallel reactions have been generated and resolved in situ by secondary lipase-catalyzed asymmetric transformation, resulting in high chemo- and enantioselectivities.
Over the last few decades, along with the powerful impact of supramolecular chemistry, the pursuit to understand the complexity of life has gained increasing momentum within the scientific community. Living systems noticeably depend on intricate networks of molecular reactions and interactions under both thermodynamic and kinetic control, operating in response to various signals and factors. Some of these features can be addressed using the concept of dynamic chemistry. Derived from the feature of reversibility, dynamic chemistry has been established as a useful methodology for generating and studying complex networks on a molecular scale. At the constitutional level, this has led to molecular networks that enable selection and identification of for example new substrates for targets, receptors for ligands, and functional materials.1–3 The concept is based on the generation of dynamic systems, in which all system components mutually undergo reversible-covalent or non-covalent interactions under thermodynamic control. The adaptive nature of these systems makes them responsive to internal or external pressures, and any changes of the important parameters in the system can rearrange the constituent compositions, leading to the amplification of optimal constituents. Among the variety of selection pressures that have been applied to dynamic systems, secondary, kinetically controlled, enzyme-catalyzed transformations have proven especially efficient in constituent selection, due to their high substrate specificities.4–10 Lipases, which belong to the hydrolase enzyme family, were most often used in the dynamic systems owing to advantages such as showing high catalytic efficiency and good selectivities, and being environmentally friendly.11–14 In these systems, not only new reversible reactions have been applied, but also the catalytic promiscuities of enzymes have been challenged, generating novel dynamic systemic resolutions with high chemo- and stereoselectivities. Within this context, however, dynamic systems have generally been based on single reversible reaction formats. The principle is certainly fully amenable to interconnected reversible reactions, generating systems of considerably higher complexity,8,15–18 but presents a challenge with respect to their generation and control. Yet, this represents an important development since higher order systems would lead to an increased understanding of molecular complexity in general (Fig. 1).
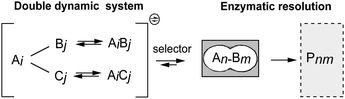 | ||
| Fig. 1 Double parallel dynamic systemic resolution. | ||
This challenge was addressed in the present study, where new dynamic systems were generated based on two different reversible reactions operating in parallel under the same conditions, providing two types of structures for the secondary enzymatic resolution process. Several requirements need to be met by the reactions operating in such systems: (1) compatibility under the same catalytic conditions; (2) similar kinetic properties with comparable substrate distributions; and (3) no dominant side reactions taking place in the system during the time course. In the present case, the nitroaldol reaction together with hemithioacetal formation were deemed able to fulfill all prerequisites, and further evaluated in a more complex parallel resolution process. The nitroaldol reaction is one of the first C–C bond-forming reactions used for complex dynamic systems, and its reversibility has been confirmed under basic conditions.6,19 Hemithioacetal formation has also been utilized in both aqueous and organic solvents,9,20,21 leading to the rapid formation of virtual dynamic systems in water, whereas base catalyzed real dynamic systems can be produced in organic solvents.
Due to the discrete properties of each of the two reaction types, the equilibration features were first addressed. Starting from equimolar amounts of 3-nitrobenzaldehyde 1, 2-nitropropane A, and 1-butanethiol B, five equivalents of base triethylamine (TEA) were added to the system to initiate the reversible nitroaldol reaction. 1H NMR spectroscopy was used to follow the dynamic system with CDCl3 as the solvent at room temperature. Not surprisingly, these two reactions showed totally different kinetics: hemithioacetal intermediate 1B was formed immediately while nitroaldol intermediate 1A was more slowly produced with time. The final equilibrium for the whole system was reached in 15 h, in which the ratio between 1A and 1B was 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, indicating that the nitroaldol intermediate is thermodynamically more stable than the hemithioacetal intermediate under these conditions. However, the comparable intermediate amounts still showed that these two reversible reactions can operate simultaneously in one system, thus being potentially applicable to the enzymatic resolution process.
:5, indicating that the nitroaldol intermediate is thermodynamically more stable than the hemithioacetal intermediate under these conditions. However, the comparable intermediate amounts still showed that these two reversible reactions can operate simultaneously in one system, thus being potentially applicable to the enzymatic resolution process.
A more complex dynamic system was subsequently generated with two additional aldehydes, 1-chlorobenzaldehyde 2 and 2,4-dichlorobenzaldehyde 3, under the same conditions (Scheme 1). The equilibrium was in this case reached within approximately the same time frame as previously (16 h), displaying the same trend of thermodynamically preferred nitroaldol intermediates where the ratios to the corresponding hemithioacetal intermediates were higher (Fig. 2b). The preference sequence of the three aldehydes for both reaction intermediates was 1 > 3 > 2, following the trend of different activation effects from electron withdrawing substituents of the aldehydes.
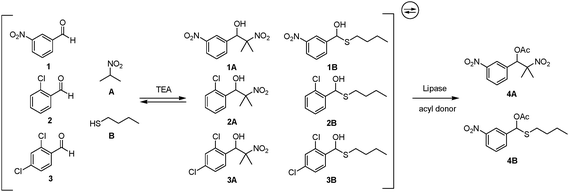 | ||
| Scheme 1 Generation of a double parallel dynamic system coupled to lipase-catalyzed asymmetric resolution. | ||
 | ||
| Fig. 2 1H-NMR spectra of a double parallel dynamic system: (a) the full spectrum of a typical dynamic system; (b) enlarged area of 16 h after generation of a dynamic system, all components in equilibrium before enzymatic resolution; (c) enlarged area of 10 d after adding lipase. | ||
After addressing the equilibrium features, the double parallel dynamic system was subjected to a secondary enzymatic resolution process. Since two different types of intermediates were present in the same system, the enzymatic activity in selective transesterification reactions was targeted. Two different lipases, Burkholderia cepacia (formerly Pseudomonas cepacia) lipase (PS-IM) and Pseudozyma antarctica (formerly Candida antarctica) lipase B (CAL-B), commonly used in organic synthesis, were chosen to test this double dynamic system.22–24 Together with phenyl acetate, one of the activated acyl donors for lipase transesterifications, these two lipases behaved totally different under the same conditions: CAL-B gave no product from either intermediates while PS-IM provided both acetylated products 4A and 4B in different amounts. However, no products from aldehyde 2 or 3 were observed, thus unselected by lipase-catalyzed transesterifications while in the same system with aldehyde 1. Between the two types of intermediate structures, hemithioacetal intermediate 1B was slightly preferred over the corresponding nitroaldol structure, which was in a converse ratio to their equilibrium distributions. Thus, PS-IM possesses a stronger preference for the hemithioacetal intermediate, leading to the hemithioacetal ester product.
In order to improve the enzyme selectivity between the nitroaldol and hemithioacetal intermediates, various parameters were subsequently explored (Table 1). Different reaction media were thus tested: toluene and tert-butyl methyl ether (TBME). The result showed that hemithioacetal product 4B was produced at a much higher rate in TBME. On the other hand, acetylation of nitroaldol intermediate 1A occurred at almost the same rate as in toluene, resulting in better total conversion and a higher ratio between 4B and 4A. Lower loading of PS-IM was also applied, 200 mg, 100 mg, and 50 mg enzyme preparation. Although the selectivity was increased with a lower amount of PS-IM, considerably lower conversions were observed as expected. Finally, different temperatures were screened: room temperature and 0 °C. Interestingly, at lower temperature, the product ratio between 4B and 4A was much higher, increasing from 4:1 to 17:1, while the total conversion was similar compared to the corresponding result at room temperature. The higher selectivity may in part be explained by the more rigid structure of PS-IM at 0 °C, resulting in a less flexible active site.25,26 Obviously conversion of this acetylation process is not very sensitive to temperature, and was not affected. Therefore with TBME as solvent at 0 °C, only trace amounts of 4A were detected in the 1H NMR spectrum, resulting in high selectivity of 4B from the double parallel dynamic system reaching a total conversion of 76% (Fig. 2c).
| Entry | Solvent | Amount of PS-IM (mg) | T (°C) | Reaction time (days) | Conversion (%) | 4B:4A ratio |
|---|---|---|---|---|---|---|
| a Reaction conditions: 0.1 mmol of each benzaldehyde 1, 2, 3, 2-nitropropane and 1-butanethiol, 0.5 mmol TEA, 0.3 mmol phenyl acetate, 0.6 mL toluene/TBME were added to the vial of PS-IM with 20 mg of 4 Å molecular sieves. The reactions were monitored by 1H NMR spectroscopy. | ||||||
| 1 | Toluene | 200 | rt | 10 | 51 | 2:1 |
| 2 | TBME | 200 | rt | 10 | 71 | 4:1 |
| 3 | TBME | 100 | rt | 11 | 63 | 5:1 |
| 4 | TBME | 50 | rt | 11 | 50 | 5:1 |
| 5 | TBME | 200 | 0 | 10 | 75 | 17:1 |
An important advantage of using enzyme-catalyzed reactions for resolution processes is its potential high stereoselectivity toward the substrates. Therefore, chiral HPLC was applied to check the enantiomeric excess (ee) of product 4B from this double dynamic system.
Very modest enantiomeric purities were however observed, with 46% ee after silica column purification. In order to improve the enantiomeric selectivity, a series of acyl donors: isopropyl acetate, isopropenyl acetate, and substituted phenyl acetates were screened; but compared to phenyl acetate, very similar results were observed. Surprisingly, when the single reaction between 1 and B, together with PS-IM-catalyzed transesterification, was tested under the same conditions, without column purification prior to HPLC analysis, a much higher ee (up to 93.3%) was detected. The lower ee from the dynamic system could thus be attributed to the product racemization under the mild acidic conditions of the silica column. To address this problem, neutral aluminium oxide based purification of product 4B was instead conducted, and the final enantiomeric purity of 4B from the complex system was recorded as 90.7% ee. Therefore, not only good chemoselectivity, but also high enantioselectivity could be achieved from this complex double parallel dynamic system.
In summary, double parallel dynamic systems have been developed, in which nitroaldol and hemithioacetal formation was mutually compatible under the same conditions, providing substrate structures with higher complexities for further transformations. Biocatalysis, lipase-catalyzed transesterification, was subsequently challenged by this parallel dynamic system as the secondary kinetic resolution process. Between the two types of substrates, the hemithioacetal intermediates were much preferred over the nitroaldol intermediates, with a single product being amplified from the system, thus resulting in high chemo- and enantioselectivity of enzymatic resolutions in the one-pot process. This methodology represents a new way to build more complex dynamic systems, with increasing potential for applications in for example more efficient substrate screening protocols and receptor–ligand recognition studies. From a long-term perspective, it furthermore sheds light on considerably more complex systems.
This work was in part supported by the Swedish Research Council and the Royal Institute of Technology. YZ and LH thank the China Scholarship Council for special scholarship awards.
Notes and references
- (a) Dynamic Combinatorial Chemistry: In Drug Discovery, Bioorganic Chemistry, and Materials Science, ed. B. L. Miller, John Wiley & Sons, Inc., Hoboken, NJ, 2010 Search PubMed; (b) Dynamic Combinatorial Chemistry, ed. J. N. H. Reek and S. Otto, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010 Search PubMed; (c) Constitutional Dynamic Chemistry, ed. M. Barboiu, Springer Verlag, Berlin Heidelberg, 2012 Search PubMed.
- (a) P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J. L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652 CrossRef CAS; (b) B. De Bruin, P. Hauwert and J. N. H. Reek, Angew. Chem., Int. Ed., 2006, 45, 2660 CrossRef CAS; (c) R. A. R. Hunt and S. Otto, Chem. Commun., 2011, 47, 847 RSC; (d) J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151 RSC; (e) J.-M. Lehn, Top. Curr. Chem., 2012, 322, 1 CrossRef CAS; (f) O. Ramström, T. Bunyapaiboonsri, S. Lohmann and J.-M. Lehn, Biochim. Biophys. Acta, Gen. Subj., 2002, 1572, 178 CrossRef; (g) O. Ramström and J.-M. Lehn, Nat. Rev. Drug Discovery, 2002, 1, 26 CrossRef.
- (a) P. Besenius, P. A. G. Cormack, R. F. Ludlow, S. Otto and D. C. Sherrington, Org. Biomol. Chem., 2010, 8, 2414 RSC; (b) A. Granzhan, C. Schouwey, T. Riis-Johannessen, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2011, 133, 7106 CrossRef CAS; (c) K. Imato, M. Nishihara, T. Kanehara, Y. Amamoto, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2012, 51, 1138 CrossRef CAS; (d) P. Kovaříček and J.-M. Lehn, J. Am. Chem. Soc., 2012, 134, 9446 CrossRef; (e) N. Roy and J.-M. Lehn, Chem.–Asian J., 2011, 6, 2419 CrossRef CAS; (f) L. Tauk, A. P. Schröder, G. Decher and N. Giuseppone, Nat. Chem., 2009, 1, 649 CrossRef CAS; (g) L. You, J. S. Berman and E. V. Anslyn, Nat. Chem., 2011, 3, 943 CrossRef CAS; (h) Y. Zhang, M. Angelin, R. Larsson, A. Albers, A. Simons and O. Ramström, Chem. Commun., 2010, 46, 8457 RSC.
- M. Sakulsombat, Y. Zhang and O. Ramström, Top. Curr. Chem., 2012, 322, 55 CrossRef CAS.
- R. Larsson, Z. Pei and O. Ramström, Angew. Chem., Int. Ed., 2004, 43, 3716 CrossRef CAS.
- R. Larsson and O. Ramström, Eur. J. Org. Chem., 2006, 285 CrossRef CAS.
- P. Vongvilai, M. Angelin, R. Larsson and O. Ramström, Angew. Chem., Int. Ed., 2007, 46, 948 CrossRef CAS.
- P. Vongvilai and O. Ramström, J. Am. Chem. Soc., 2009, 131, 14419 CrossRef CAS.
- M. Sakulsombat, P. Vongvilai and O. Ramström, Org. Biomol. Chem., 2011, 9, 1112 CAS.
- M. Sakulsombat, Y. Zhang and O. Ramström, Chem.–Eur. J., 2012, 18, 6129 CrossRef CAS.
- M. Breuer, K. Ditrich, T. Habicher, B. Hauer, M. Keβeler, R. Stürmer and T. Zelinski, Angew. Chem., Int. Ed., 2004, 43, 788 CrossRef CAS.
- H. E. Schoemaker, D. Mink and M. G. Wubbolts, Science, 2003, 299, 1694 CrossRef CAS.
- M. Kapoor and M. N. Munishwar, Process Biochem., 2012, 47, 555 CrossRef CAS.
- A. Houde, A. Kademi and D. Leblanc, Appl. Biochem. Biotechnol., 2004, 118, 155 CrossRef CAS.
- N. Giuseppone, J.-L. Schmitt, E. Schwartz and J.-M. Lehn, J. Am. Chem. Soc., 2005, 127, 5528 CrossRef CAS.
- J. Leclaire, L. Vial, S. Otto and J. K. M. Sanders, Chem. Commun., 2005, 1959 RSC.
- M. Barboiu, E. Petit, A. van der Lee and G. Vaughan, Inorg. Chem., 2005, 45, 484 CrossRef.
- D. G. Hamilton, N. Feeder, S. J. Teat and J. K. M. Sanders, New J. Chem., 1998, 22, 1019 RSC.
- P. Vongvilai, R. Larsson and O. Ramström, Adv. Synth. Catal., 2008, 350, 448 CrossRef CAS.
- R. Caraballo, H. Dong, J. P. Ribeiro, J. Jiménez-Barbero and O. Ramström, Angew. Chem., Int. Ed., 2010, 49, 589 CrossRef CAS.
- R. Caraballo, M. Sakulsombat and O. Ramström, ChemBioChem, 2010, 11, 1600 CrossRef CAS.
- G. Soberon-Chavez and B. Palmeros, Crit. Rev. Microbiol., 1994, 20, 95 CrossRef CAS.
- F. Secundo and G. Carrea, J. Mol. Catal. B: Enzym., 2002, 19–20, 93 CrossRef CAS.
- O. Pàmies and J.-E. Bäckvall, Trends Biotechnol., 2004, 22, 130 CrossRef.
- P. L. A. Overbeeke, J. Ottosson, K. Hult, J. A. Jongejan and J. A. Duine, Biocatal. Biotransform., 1999, 17, 61 CrossRef CAS.
- T. Sakai, I. Kawabata, T. Kishimoto, T. Ema and M. Utaka, J. Org. Chem., 1997, 62, 4906 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and NMR spectra. See DOI: 10.1039/c3cc38203f |
| This journal is © The Royal Society of Chemistry 2013 |