 Open Access Article
Open Access ArticlePositively charged bulk Au particles as an efficient catalyst for oxidation of styrene with molecular oxygen†
Liang
Wang
b,
Bingsen
Zhang
c,
Wei
Zhang
d,
Jian
Zhang
a,
Xionghou
Gao
e,
Xiangju
Meng
a,
Dang Sheng
Su
*cd and
Feng-Shou
Xiao
*a
aKey Laboratory of Applied Chemistry of Zhejiang Province, Zhejiang University, Hangzhou 310028, China. E-mail: fsxiao@zju.edu.cn
bState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, Jilin University, Changchun 130012, China
cShenyang National Laboratory of Materials Science, Institute of Metal Research, Chinese Academy of Science, Shenyang 110016, China. E-mail: dangsheng@fhi-berlin.mpg.de
dDepartment of Inorganic Chemistry, Fritz Haber Institute of the Max Planck Society, Berlin 14195, Germany
eLanzhou Petrochemical Research Center, Petrochemical Research Institute, Petro China Company Limited, Lanzhou 730060, China
First published on 7th March 2013
Abstract
Positively charged bulk Au particles with size of 20–150 nm were formed on amino-modified porous polydivinylbenzene (Au/PN), which showed superior catalytic performance and good recyclability in the aerobic oxidation of styrene.
Since the pioneering work of Hutchings and Haruta et al.,1 Au catalysts have been demonstrated to be active for many important reactions, especially for oxidation by molecular oxygen,2–7 where a critical factor for determining the catalytic performance is the particle size of the Au catalysts. When the Au particle sizes are over 5 nm, the Au catalysts are normally of low-efficiency.4a,6,8
As an important model reaction for the production of fine chemicals and pharmaceuticals, the oxidation of styrene has been widely investigated over various Au catalysts.2,9–14 For the liquid-phase oxidation of styrene by molecular oxygen, additives (e.g. t-butyl hydroperoxide) as the initiator are necessary.2,11,12 However, when the size of Au particles is extremely small (∼1.4 nm, 55 atoms, or ∼0.9 nm, 25 atoms), the Au catalysts are very active even in the absence of additives.9,10 In contrast, when the size of Au particles is 2 nm, they become completely inactive. Notably, the synthesis of extremely small Au nanoparticles usually requires precursor ligands in the starting step and the removal of the precursor ligands in the final step, which is expensive and could produce undesirable waste.8b,9,10 In addition, extremely small Au nanoparticles are unstable in catalytic oxidations due to their aggregation. As a result, their catalytic activity reduces significantly for long reactions,9 which strongly limits the wide application of Au nanoparticle catalysts with extremely small particle size.
Here, we demonstrate an alternative catalyst of amino functionalized, porous polydivinylbenzene supported, positively charged bulk Au particles (Au/PN) with particle sizes of 20–150 nm, which are very stable and active in the aerobic oxidation of styrene. This result is quite different from the known fact that only Au particles with a size smaller than 2 nm are catalytically active and stable for the aerobic oxidation of styrene in the absence of initiators.9,15
The Au/PN sample is synthesized from the one-pot treatment of amino functionalized porous polydivinylbenzene with HAuCl4 solution. The choice of amino groups is due to our careful investigation of the interaction between the HAuCl4 solution with various amines. Fig. S1 (ESI†) shows the photographs and UV-Visible spectra of mixtures of the HAuCl4 solution with different amines. When the aliphatic amines cyclohexylamine and ethylenediamine were added into the HAuCl4 solution, the mixture still exhibited a yellow color, which was very similar to the color of the pure HAuCl4 solution (Fig. S1A, ESI†). In addition, the mixture of cyclohexylamine and HAuCl4 exhibited a similar UV-Visible spectra (Fig. S1B-c, ESI†) to the pure HAuCl4 (Fig. S1B-a, ESI†), indicating that the valence of Au cations was stable in the mixture of cyclohexylamine with HAuCl4. Interestingly, when aniline was added into the HAuCl4 solution, the color immediately changed to dark brown. In comparison with the UV-Vis spectra of pure HAuCl4 solution (Fig. S1B-a, ESI†) and pure aniline (Fig. S1B-d, ESI†), the dark brown mixture showed clear additional signals in the wavelength of 500–700 nm (Fig. S1B-e, ESI†), which are associated with the presence of metallic Au.16 These results indicate that metallic Au species could be formed by the interaction between aniline and Au cations.
Based on the interaction between aniline and Au cations, it is suggested that amino-modified porous polydivinylbenzene (PN) should be a suitable support for the formation of metallic Au species as a heterogeneous catalyst (Au/PN). Fig. 1A shows the synthesis scheme of Au/PN where the Au content is about 2.4 wt% (Table S1, ESI†).
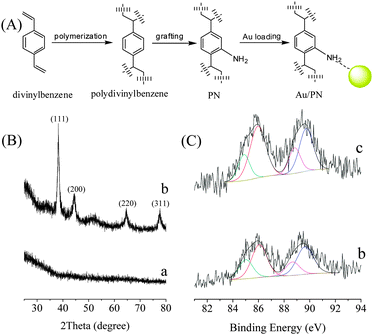 | ||
| Fig. 1 (A) Scheme, (B) XRD, and (C) Au4f XPS spectra of (a) PN, (b) Au/PN, and (c) Au/PN-R. | ||
The presence of amino groups on polydivinylbenzene was confirmed by IR and N1s XPS techniques (Fig. S2, ESI†). In addition, the XRD pattern of Au/PN shows four obvious peaks at 38.3, 44.5, 64.7, and 77.7°, which are attributed to the (111), (200), (220), and (311) incidences of a typical metallic Au species (Fig. 1B). Notably, these peaks are narrow and sharp, suggesting a relatively large particle size of metallic Au.16 Furthermore, the UV-Vis spectrum of Au/PN gives an obvious peak at 518 nm (Fig. S3, ESI†), confirming the presence of metallic Au species in the Au/PN sample,16 in good agreement with the results of the XRD patterns (Fig. 1B).
Fig. 2 shows the microscopic characterization of Au/PN. In the STEM image, the Au particles with large sizes of 20–150 nm are clearly observed on the porous PN support (Fig. 2a and b). The elemental mapping shows that the C and N species are homogeneously distributed on the Au/PN sample (Fig. 2c and d). Notably, the distribution of Au particles in the STEM image (the white square in Fig. 2a) is highly consistent with the distribution of Au species in the Au elemental mapping (Fig. 2e). A similar result could also be found in various areas of the Au/PN sample (Fig. S4, ESI†), indicating that the Au species on the Au/PN are present as bulk Au particles. Additionally, the HRTEM images show that extremely small Au nanoparticles could not be detected on the Au/PN (Fig. S5, ESI†), which supports that only bulk Au particles exist in the Au/PN sample.
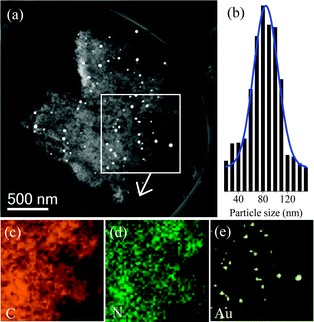 | ||
| Fig. 2 (a) STEM image, (b) particle size distribution, (c–e) C, N, and Au elemental maps of Au/PN. | ||
Although the bulk metallic Au particles have been confirmed on the Au/PN sample, it is still desirable to investigate the electronic properties of the Au species on the surface of the bulk Au. Fig. 1C shows the Au4f XPS spectra of various samples. Interestingly, the Au4f XPS spectrum of Au/PN shows obvious Au4f7/2 peaks at 86.0 and 84.9 eV associated with typical Au3+ and Au+ species, respectively.17 These results indicate that there are positively charged Au species on the surface of the bulk Au particles (Table S1, ESI†). The EPR spectra show that PN gives the signal at g = 2.0045, while Au/PN has an additional strong signal (g = 2.3262), which is reasonably assigned to the positively charged Au species (Fig. S6, ESI†) because the pure metallic Au is EPR-silent.18 In combination of the results obtained from the XPS and EPR techniques, we could conclude that the bulk Au particles of Au/PN are rich in Au cations. Very surprisingly, the Au cations on Au/PN are extremely stable. For example, after reduction of the Au/PN sample by NaBH4, the obtained Au/PN-R still shows similar Au4f XPS (Fig. 1C-c, Table S1, ESI†) and EPR spectra (Fig. S6-c, ESI†) to those of the as-synthesized Au/PN. These results indicate that the Au cations on Au/PN have extraordinarily high stability, which might be related to the interaction between the aniline groups and the Au species.
For comparison, the aliphatic amino functionalized mesoporous silica was synthesized by coating aminopropyltriethoxysilane (KH-550) on the surface of MCM-41 (MCM-N, Scheme S1, ESI†). When the Au species were loaded on MCM-N (Au/MCM-N), the metallic Au nanoparticles were undetectable from the XRD pattern (Fig. S7, ESI†). After reduction with NaBH4, Au3+ cations were undetectable on the Au/MCM-N sample, indicating that Au3+ cations are unstable in the presence of NaBH4 (Fig. S7–S9, ESI†), which is quite distinguishable from the phenomenon of the Au/PN sample with very stable Au cations.
Table 1 presents the catalytic data of various Au catalysts in the direct aerobic oxidation of styrene. The PN support is completely inactive for this reaction (entry 1 in Table 1). However, the Au/PN catalyst shows high styrene conversion at 27.0%, with selectivity towards styrene epoxide at 30% (entry 2 in Table 1, Fig. S12, ESI†). In comparison, the non-porous supported Au particles (Au/non-porous PN, Table S1, Fig. S10 and S11, ESI†) with similar Au particle sizes and cations, show styrene conversion at 14.2% (entry 5 in Table 1, Table S2, ESI†). The extremely small Au nanoparticles (∼1.4 nm) supported on boron nitride (Au55/BN) and SiO2 (Au55/SiO2), which were reported as very efficient catalysts for the aerobic oxidation of styrene, show styrene conversion at 19.2 and 25.8, with styrene epoxide selectivity at 12–14% (entries 6 and 7 in Table 1). These results indicate that the Au/PN catalyst is very active (Fig. S13, ESI†).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Conv. (%) | Product selectivityb (%) | Ref. | |||
| P1 | P2 | P3 | P4 | ||||
| a Reaction conditions: 100 mg of catalyst, 12 mmol of styrene, 20 mL of toluene, 1.5 bar of O2, 100 °C, 15 h, dodecane as the internal standard. b P1 = styrene epoxide, P2 = benzaldehyde, P3 = acetophenone, P4 = phenylacetaldehyde, benzyl alcohol and others. c 1.0 bar of O2. d 25 mg of catalyst. e 1.3 mg of catalyst. | |||||||
| 1 | PN | Inactive | This work | ||||
| 2 | Au/PN | 27.0 | 30.0 | 65.2 | 2.8 | 2.0 | This work |
| 3c | Au/PN | 16.4 | 22.2 | 67.2 | 10.6 | — | This work |
| 4d | Au/PN | 24.9 | 31.2 | 64.8 | 2.1 | 1.9 | This work |
| 5 | Au/non-porous PN | 14.2 | 29.1 | 60.2 | 3.3 | 7.4 | This work |
| 6 | Au55/BN | 19.2 | 14.0 | 82.3 | 3.9 | 9 | |
| 7 | Au55/SiO2 | 25.8 | 12.0 | 82.1 | 5.7 | 9 | |
| 8 | Au/MCM-N-R | Inactive | This work | ||||
| 9 | Au/MCM-N | Inactive | This work | ||||
| 10e | HAuCl4 | Inactive | This work | ||||
| 11 | Au/PN-R | 27.1 | 29.0 | 65.7 | 3.1 | 2.2 | This work |
In contrast, the Au/MCM-N-R catalyst with small Au nanoparticles of 2.0–3.4 nm (Fig. S9 and Table S1, ESI†) is completely inactive for this reaction (entry 8 in Table 1).9 Additionally, both the Au/MCM-N and HAuCl4 catalysts, which have only Au3+ cations, are also inactive for this reaction (entries 9 and 10 in Table 1). These results might suggest that the Au0 on the Au particles (>2 nm) or Au3+ sites are not the active centres for the aerobic oxidation of styrene. Based on these results, it might be suggested that the Au+ sites on Au/PN are the active sites for the oxidation of styrene, because the Au+ sites have similar Au4f7/2 electron binding energy (85.0 eV) to that of the Au55 clusters (85.1 eV), suggesting their similar electronic properties which might lead to unusual catalytic performances.9,17,19
It is very important to point out that there is no leaching for the Au/PN catalyst. For example, after filtrating the used Au/PN catalyst, the Au concentration in the liquor is less than 3 ppb (ICP analysis). More importantly, Au/PN has very excellent recyclability. Even when recycled 7 times, the sample still exhibits very similar activities (Fig. 3). On the contrary, the Au55/SiO2 catalyst loses most of its activity after two further cycles under the same reaction conditions (Fig. 3), because the Au55 clusters are not stable and aggregate into relatively large nanoparticles during the reaction process.9
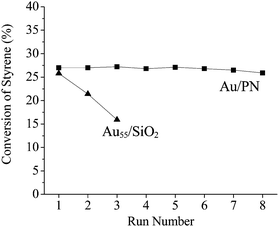 | ||
| Fig. 3 Aerobic oxidation of styrene using Au/PN and Au55/SiO2 catalysts over several cycles. The data for the Au55/SiO2 catalyst is from ref. 9. | ||
This work is supported by the National Natural Science Foundation of China (20973079 and U1162201) and State Basic Research Project of China (2009CB623501). Bingsen Zhang gratefully acknowledge the financial support provided by the IMR SYNL-T.S. Kê Research Fellowship, the National Natural Science Foundation of China (No. 21203215), and the China Postdoctoral Science Foundation (2012M520652).
Notes and references
- (a) G. J. Hutchings, J. Catal., 1985, 96, 292 CrossRef CAS; (b) M. Haruta, T. Koboyashi, H. Sano and N. Yamada, Catal. Lett., 1987, 16, 405 Search PubMed.
- M. D. Hughes, Y.-J. Xu, P. Jenkins, P. McMorn, P. Landon, D. I. Enache, A. F. Carley, G. A. Attard, G. J. Hutchings, F. King, E. H. Stitt, P. Johnston, K. Griffin and C. J. Kiely, Nature, 2005, 437, 1132 CrossRef CAS.
- (a) A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096 RSC; (b) D. P. He, H. Shi and B.-Q. Xu, Green Chem., 2007, 9, 849 RSC; (c) F.-Z. Su, Y.-M. Liu, L.-C. Wang, Y. Cao, H.-Y. He and K.-N. Fan, Angew. Chem., Int. Ed., 2008, 47, 334 CrossRef CAS.
- (a) A. Abad, P. Concepcion, A. Corma and H. Garcia, Angew. Chem., Int. Ed., 2005, 44, 4066 CrossRef CAS; (b) J. C. F. Gonzales and B. C. Gates, Chem. Soc. Rev., 2008, 37, 2127 RSC.
- (a) X. Zhang, H. Shi and B.-Q. Xu, Angew. Chem., Int. Ed., 2005, 44, 7132 CrossRef CAS; (b) C. Y. Ma, Z. Mu, J. J. Li, Y. G. Jin, J. Cheng, G. Q. Lu, Z. P. Hao and S. Z. Qiao, J. Am. Chem. Soc., 2010, 132, 2608 CrossRef CAS; (c) W. Yan, S. Brown, Z. Pan, S. M. Mahurin, S. H. Overbury and S. Dai, Angew. Chem., Int. Ed., 2006, 45, 3614 CrossRef CAS.
- S. Abraham, I. Kim and C. A. Batt, Angew. Chem., Int. Ed., 2007, 46, 5720 CrossRef CAS.
- (a) L. Wang, W. Zhang, D. S. Su, X. Meng and F.-S. Xiao, Chem. Commun., 2012, 48, 5476 RSC; (b) P. Liu, Y. Guan, R. A. van Santen, C. Li and E. J. M. Hensen, Chem. Commun., 2011, 47, 11540 RSC.
- (a) M. Stratakis and H. Garcia, J. Am. Chem. Soc., 2012, 112, 4469 CAS; (b) H. Tsunoyama, N. Ichikuni, H. Sakuria and T. Tsukuda, J. Am. Chem. Soc., 2009, 131, 7086 CrossRef CAS.
- M. Turner, V. B. Golovko, O. P. H. Vaughan, P. Abdulkin, A. Berenguer-Murcia, M. S. Tikhov, B. F. G. Johnson and R. M. Lambert, Nature, 2008, 454, 981 CrossRef CAS.
- Y. M. Liu, H. Tsunoyama, T. Akita and T. Tsukuda, Chem. Commun., 2010, 46, 550 RSC.
- L. Wang, H. Wang, P. Hapala, L. Zhu, L. Ren, X. Meng, J. P. Lewis and F.-S. Xiao, J. Catal., 2011, 281, 30 CrossRef CAS.
- F.-X. Zhu, W. Wang and H.-X. Li, J. Am. Chem. Soc., 2011, 133, 11632 CrossRef CAS.
- X. Deng and C. M. Friend, J. Am. Chem. Soc., 2005, 127, 17178 CrossRef CAS.
- Y. Zhu, H. Qian, M. Zhu and R. Jin, Adv. Mater., 2010, 22, 1915 CrossRef CAS.
- H. G. Boyen, G. Kastle, F. Weigl, B. Koslowaki, C. Dietrich, P. Ziemann, J. P. Spatz, S. Riethmuller, C. Hartmann, M. Moller, G. Schmid, M. G. Garnier and P. Oelhafen, Science, 2002, 297, 1533 CrossRef CAS.
- L. Wang, X. Meng, B. Wang, W. Chi and F.-S. Xiao, Chem. Commun., 2010, 46, 5003 RSC.
- R. Si and M. Flytzani-Stephanopoulos, Angew. Chem., Int. Ed., 2008, 47, 2884 CrossRef CAS.
- B. Chowdhury, J. J. Bravo-Suarez, N. Mimura, J. Lu, K. K. Bando, S. Tsubota and M. Haruta, J. Phys. Chem. B, 2006, 110, 22995 CrossRef CAS.
- L. K. Ono, D. Sudfeld and B. R. Cuenya, Surf. Sci., 2006, 600, 5041 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cc00309d |
| This journal is © The Royal Society of Chemistry 2013 |