 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of selective inhibitors of sphingosine kinase 1†
Dong Jae
Baek
a,
Neil
MacRitchie
b,
Nigel J.
Pyne
b,
Susan
Pyne
b and
Robert
Bittman
*a
aDepartment of Chemistry and Biochemistry, Queens College of the City University of New York, Flushing, New York 11367-1597, USA. E-mail: robert.bittman@qc.cuny.edu; Tel: +1 718-997-3279
bCell Biology Group, Strathclyde Institute of Pharmacy and Biomedical Sciences, University of Strathclyde, Glasgow G4 0RE, UK
First published on 28th January 2013
Abstract
Sphingosine kinase isoform 1 (SK1) inhibitors may serve as therapeutic agents for proliferative diseases, including hypertension. We synthesized a series of sphingosine-based SK1-selective inhibitors, the most potent of which is RB-005 (IC50 = 3.6 μM), which also induced proteasomal degradation of SK1 in human pulmonary arterial smooth muscle cells.
S1P is formed from sphingosine by two isoforms of sphingosine kinase (SK1 and SK2) that are encoded by distinct genes and differ in their biochemical properties, subcellular localization, and function.1 Binding of S1P to 5 cell-surface G-protein coupled S1P receptors (termed S1P1–5) induces a multitude of cell responses. S1P also has intracellular targets.2–4 Targeting SK1 is also of particular relevance to cancer.1 Many human tumor types exhibit increased levels of SK1 mRNA and/or protein which correlate, in several studies, with increased tumor grade, reduced patient survival, and development of chemotherapeutic resistance to anti-cancer agents. Indeed, knockdown of SK1 reduces proliferation of glioblastoma cells5 and androgen-independent PC-3 prostate cancer cells.6
We have demonstrated that sustained hypoxia induces an increase in the transcription of the SK1 gene in proliferating human pulmonary arterial smooth muscle cells (PASMC) that might account for their increased survival7 and thereby contribute to vascular remodeling in pulmonary arterial hypertension (PAH). Moreover, others have reported lower levels of ceramide in vascular smooth muscle cells isolated from spontaneously hypertensive rats,8 suggesting that hypertension is associated with increased conversion of ceramide to sphingosine and S1P. Here, we report the synthesis and evaluation of new sphingosine-based SK1-selective inhibitors, which may have potential as inhibitors of proliferative diseases, including PAH.
Several SK inhibitors have been previously identified. For example, the non-sphingoid molecule 2-(p-hydroxyanilino)-4-(p-chlorophenyl)thiazole (SKi, also referred to as SKI-II; see Fig. 1) reduced intracellular S1P, inhibited proliferation, and induced apoptosis in various cancer cell lines.9 SKi is orally bioavailable, inhibits tumor growth10 and ulceritive colitis,11 and reduces airway hyper-responsiveness.12 The sphingosine analogue (2R,3S,4E)-N-methyl-5-(4′-pentylphenyl)-2-aminopent-4-ene-1,3-diol (BML-258, also referred to as SK1-I; Fig. 1) is a SK1-selective inhibitor.13
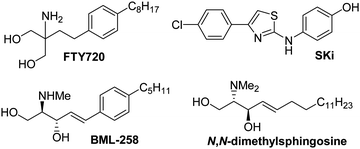 | ||
| Fig. 1 Structures of SK inhibitors. | ||
Inhibition of SK activity in cells with the non-selective isoform inhibitors SKi, N,N-dimethylsphingosine, and FTY720 (Fig. 1) induces proteasomal degradation and removal of SK1 from PASMC and cancer cell lines.14–16 Removal of SK1 reduces intracellular S1P and increases C22:0-ceramide levels, thereby promoting apoptosis.14
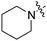
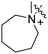
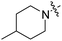
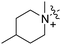
Scheme 1 outlines our synthetic route from 4-octylphenethyl alcohol (1, see Scheme S1, ESI†) to a series of FTY720-like analogues. The tertiary amines shown in Table 1 were prepared in good yields by displacement of mesylate ion from 2 with amines in acetonitrile. Some of the quaternary ammonium salts were prepared by N-alkylation of the tertiary amines (and the secondary amine RB-006) with an excess of MeI and K2CO3 in acetonitrile (Scheme S2, ESI†), whereas others (RB-011, RB-012, RB-017, and RB-018) were prepared by N-alkylation of 2 with tertiary N-methylamines (see ESI†).
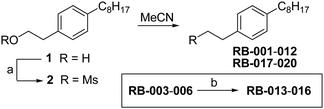 | ||
| Scheme 1 Synthesis of tertiary amine derivatives (RB-001–010, 019 and 020) and quaternary ammonium salts (RB-011–018) of FTY720. (a) MsCl, Et3N, CH2Cl2; (b) MeI, K2CO3, MeCN. | ||
| Compound | R | Compound | R |
|---|---|---|---|
| RB-001 |
|
RB-011 |

|
| RB-002 |
|
RB-012 |

|
| RB-003 |

|
RB-013 |
|
| RB-004 |
|
RB-014 |
|
| RB-005 |

|
RB-015 |

|
| RB-006 |
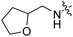
|
RB-016 |

|
| RB-007 |
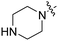
|
RB-017 |
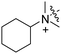
|
| RB-008 |
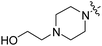
|
RB-018 |
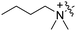
|
| RB-009 |

|
RB-019 |

|
| RB-010 |
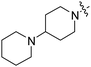
|
RB-020 |

|
We have established SK1 and SK2 assays using sphingosine/[32P]-ATP with over-expressed recombinant SK1 in HEK 293 cell lysates or purified SK2.15,16 By using sphingosine concentrations of 3 μM and 10 μM, which correspond to the Km values of SK116 and SK2,17 respectively, we demonstrate here that RB-001–003, RB-005, RB-007–009, RB-019 (Table 1), and RB-021 (Scheme 2) are selective inhibitors of SK1 over SK2.
The data in Fig. 2 show that the lead molecules for further structural optimization are 1-(4-octylphenethyl)piperidin-4-ol (RB-005) and 1-(4-octylphenethyl)piperidin-3-ol (RB-019) because these compounds have the highest selectivity for SK1 over SK2 (15.0- and 6.1-fold, respectively). These compounds feature an n-octylphenyl group linked in a 2-carbon tether to the nitrogen of 4-hydroxy- or 3-hydroxypiperidine, respectively. Small changes in the structure of this tertiary amine result in large changes in selectivity, arguing for the hypothesis that RB-005 is a highly selective inhibitor of SK1. The hydroxyl group in the heterocyclic ring is important for inhibition of SK1 by RB-005. Replacement of the 4-OH group with a 4-methyl group affords RB-004, which is a moderate inhibitor of both SK1 and SK2; the 3-OH regioisomer RB-019 is a selective but less potent SK1 inhibitor. The size of the heterocycle is important as RB-003 is less selective than RB-005. The hydrophobic alkyl chain is also a key feature as sulfonation18 in this region reduces inhibition of SK1; the sulfones RB-021 and RB-022 were prepared by Sonogashira reaction of alkyne 3 with 2-(4-bromophenyl)-ethanol, followed by catalytic hydrogenation and mesylation of 4, and finally reaction with piperidine or its 4-hydroxy derivative (Scheme 2 and Scheme S3, ESI†). Conversion of the heterocyclic amines RB-003–RB-006 by N-methylation to the corresponding quaternary ammonium compounds, RB-013–RB-016, reduces inhibition of both SK1 and SK2 (Fig. 2). Relocation of the quaternary nitrogen functionality to an exocyclic position as in RB-016–RB-018 also afforded nonselective SK inhibitors (Fig. 2). This contrasts with a study that suggested that quaternary ammonium compounds are selective inhibitors of SK2 over SK1.19 The length of the intervening C–C link between the benzene ring and heterocyclic ring remains to be examined.
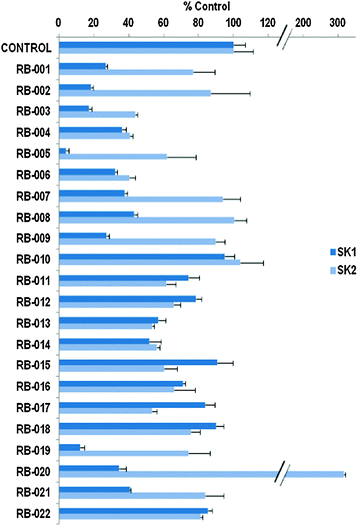 | ||
| Fig. 2 Effect of inhibitors on SK1 or SK2 activity. SK1 activity was measured using 3 μM sphingosine and 250 μM ATP. SK2 activity was assayed using 10 μM sphingosine and 250 μM ATP (n = 3 for each compound, results expressed as mean inhibition ± SD). RB series compounds were used at 50 μM. BML-258 (50 μM) inhibited SK1 activity by 74.5 ± 3.3% (n = 3). The control is 100% and equals activity against Sph alone. | ||
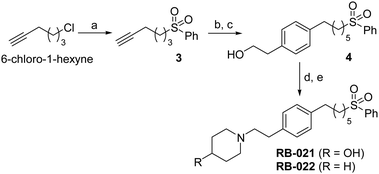 | ||
| Scheme 2 Synthesis of phenylsulfone analogues of RB-021 and RB-022. (a) PhSO2Na, THF, DMF; (b) 2-(4-bromophenyl)-ethanol, Pd(PPh3)4, CuI, Et3N; (c) 10% Pd/C, H2, EtOAc; (d) MsCl, Et3N, CH2Cl2; (e) piperidine or 4-OH-piperidine, MeCN. | ||
Next, the possibility that the six inhibitors bearing a hydroxyl group may also serve as SK substrates was examined. At 50 μM, none of these compounds are SK substrates, with the exception of RB-020, which is a weak substrate of SK1 but a good substrate of SK2 (Table 2), and RB-019, which is a very weak substrate of SK2 (<10% of Sph, data not shown).
| SK1 activity (% control) | SK2 activity (% control) | |
|---|---|---|
| a Sph was used at 3 and 10 μM for SK1 and SK2, respectively. Results are represented as % activities ± SD (n = 3 assays) of control (where 100% equals activity against Sph alone). | ||
| Sph | 100 ± 4.4 | 100 ± 12.8 |
| Sph + RB-020 | 34.4 ± 4.1 | 315 ± 5.1 |
| RB-020 | 27.5 ± 3.0 | 228 ± 17.9 |
RB-020 is less efficiently phosphorylated by SK1 than sphingosine, and probably overlaps the catalytic site of SK1 to inhibit phosphorylation of sphingosine (Table 2). However, this is not the case for SK2 where phosphorylation of sphingosine and RB-020 appear mutually exclusive (Table 2). RB-019 also inhibits SK1 activity against sphingosine (Fig. 2). Both RB-019 and RB-020 contain a hydroxyl group that is likely to be phosphorylated by SK (Table 2). It is noteworthy that RB-020 has a primary hydroxyl group attached to the heterocyclic ring through a 4-CH2 group, while RB-019 has a secondary hydroxyl group directly attached to C-3 of the heterocyclic ring; therefore, the latter hydroxyl group may be too far removed from the catalytic determinants of SK1 to be phosphorylated, but probably overlaps the substrate binding site to inhibit SK1 activity. RB-008 and RB-009 have a second N atom in the heterocyclic ring, and also possess a –(CH2)2OH and –(CH2)3OH group, respectively. However, RB-008 and RB-009 are not substrates for SK1 or SK2 (data not shown) but are inhibitors of SK1 (Fig. 2).
RB-005 inhibited SK1 activity with an IC50 value of 3.6 ± 0.38 μM (Fig. 3A). Finally, to examine whether RB-005 may function as more than a reversible inhibitor of SK1 activity, we assessed its ability to promote the proteasomal degradation of SK1 in cells.
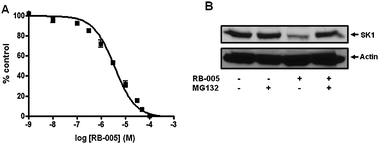 | ||
| Fig. 3 Effect of RB-005 on SK1 activity and expression. (A) Concentration-dependent inhibition of SK1 activity by RB-005 using 3 μM sphingosine and 250 μM ATP (results expressed as mean inhibition ± SD of control) (n = 3, the control is 100% and equals activity against sphingosine alone). (B) Effects of RB-005 on SK1 down-regulation. PASMC were treated with or without MG132 (10 μM, 30 min) before RB-005 (10 μM, 24 h). Cell lysates were western blotted with anti-SK1 and -actin antibodies. Results are representative of three experiments. | ||
Importantly, we found that treatment of PASMC with RB-005 (10 μM, 24 h) reduced the expression of SK1. In common with (S)-FTY720 vinylphosphonate,16 the effect of RB-005 on SK1 expression was reversed by pre-treatment of the cells with the proteasomal inhibitor MG132 (Fig. 3B).
We conclude that the FTY720 scaffold can produce selective SK1 inhibitors with low μM potency and which also induce the proteasomal degradation of SK1 in cells. Therefore, RB-005 represents a molecule for optimization to produce effective therapeutic agents.
This work was supported by a British Heart Foundation grant (29476) to NJP/SP and by NIH Grant HL-083187 to RB.
Notes and references
- N. J. Pyne and S. Pyne, Nat. Rev. Cancer, 2010, 10, 489 CrossRef CAS.
- N. C. Hait, J. Allegood, M. Maceyka, G. M. Strub, K. B. Harikumar, S. K. Singh, C. Luo, R. Marmorstein, T. Kordula, S. Milstien and S. Spiegel, Science, 2009, 325, 1254 CrossRef CAS.
- S. E. Alvarez, K. B. Harikumar, N. C. Hait, J. Allegood, G. M. Strub, E. Y. Kim, M. Maceyka, H. Jiang, C. Luo, T. Kordula, S. Milstien and S. Spiegel, Nature, 2010, 465, 1084 CrossRef CAS.
- G. M. Strub, M. Paillard, J. Liang, L. Gomez, J. C. Allegood, N. C. Hait, M. Maceyka, M. M. Price, Q. Chen, D. C. Simpson, T. Kordula, S. Milstien, E. J. Lesnefsky and S. Spiegel, FASEB J., 2011, 25, 600 CrossRef CAS.
- J. R. Van Brocklyn, C. A. Jackson, D. K. Pearl, M. S. Kotur, P. J. Snyder and T. W. Prior, J. Neuropathol. Exp. Neurol., 2005, 64, 695 CrossRef CAS.
- Y. Akao, Y. Banno, Y. Nakagawa, N. Hasegawa, T. J. Kim, T. Murate, Y. Igarashi and Y. Nozawa, Biochem. Biophys. Res. Commun., 2006, 342, 1284 CrossRef CAS.
- M. Ahmad, J. S. Long, N. J. Pyne and S. Pyne, Prostaglandins Other Lipid Mediators, 2006, 79, 278 CrossRef CAS.
- D. G. Johns, R. C. Webb and J. R. Charpie, J. Hypertens., 2001, 19, 63 CrossRef CAS.
- K. J. French, R. S. Schrecengost, B. D. Lee, Y. Zhuang, S. N. Smith, J. L. Eberly, J. K. Yun and C. D. Smith, Cancer Res., 2003, 63, 5962 CAS.
- K. J. French, J. J. Upson, S. N. Keller, Y. Zhuang, J. K. Yun and C. D. Smith, J. Pharmacol. Exp. Ther., 2006, 318, 596 CrossRef CAS.
- L. W. Maines, L. R. Fitzpatrick, K. J. French, Y. Zhuang, Z. Xia, S. N. Keller, J. J. Upson and C. D. Smith, Dig. Dis Sci., 2008, 53, 997 CrossRef CAS.
- Y. Chiba, H. Takeuchi, H. Sakai and M. Misawa, J. Pharmacol. Sci., 2010, 114, 304 CrossRef CAS.
- S. W. Paugh, B. S. Paugh, M. Rahmani, D. Kapitonov, T. Kordula, S. Milstien, J. K. Adams, R. E. Zipkin, S. Grant and S. Spiegel, Blood, 2008, 112, 1382 CrossRef CAS.
- C. Loveridge, F. Tonelli, T. Leclercq, K. G. Lim, S. Long, E. Berdyshev, R. J. Tate, V. Natarajan, S. M. Pitson, N. J. Pyne and S. Pyne, J. Biol. Chem., 2010, 285, 38841 CrossRef CAS.
- F. Tonelli, K. G. Lim, C. Loveridge, J. Long, S. M. Pitson, G. Tigyi, R. Bittman, S. Pyne and N. J. Pyne, Cell. Signalling, 2010, 22, 1536 CrossRef CAS.
- K. G. Lim, F. Tonelli, Z. Li, X. Lu, R. Bittman, S. Pyne and N. J. Pyne, J. Biol. Chem., 2011, 286, 18633 CrossRef CAS.
- K. G. Lim, S. Chaode, R. Bittman, N. J. Pyne and S. Pyne, Cell. Signalling, 2011, 23, 1590 CrossRef CAS.
- A non-lipid based phenylsulfone (PF-543) was recently reported to be a potent SK1 inhibitor: M. E. Schnute, M. D. McReynolds, T. Kasten, M. Yates, G. Jerome, J. W. Rains, T. Hall, J. Chrencik, M. Kraus, C. N. Cronin, M. Saabye, M. K. Highkin, R. Broadus, S. Ogawa, K. Cukyne, L. E. Zawadzke, V. Peterkin, K. Iyanar, J. A. Scholten, J. Wendling, H. Fujiwara, O. Nemirovskiy, A. J. Wittwer and M. M. Nagiec, Biochem. J., 2012, 444, 79 CrossRef CAS.
- M. R. Raje, K. Knott, Y. Kharel, P. Bissel, K. R. Lynch and W. L. Santos, Bioorg. Med. Chem., 2012, 20, 183 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures, NMR and mass spectra, enzyme assay procedures, and hPASMC results. See DOI: 10.1039/c3cc00181d |
| This journal is © The Royal Society of Chemistry 2013 |