 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Steric effects on excimer formation for photoluminescent smectic liquid-crystalline materials†
Shogo
Yamane
,
Yoshimitsu
Sagara
and
Takashi
Kato
*
Department of Chemistry and Biotechnology, School of Engineering, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: kato@chiral.t.u-tokyo.ac.jp
First published on 6th February 2013
Abstract
The introduction of bulky tert-butyl substituents into smectic liquid-crystalline 9,10-bis(phenylethynyl)anthracene derivatives influences excimer structures and luminescence properties.
Anthracene is an acene compound that has been widely investigated for its potential applications in semiconductors1 and luminescent materials.2–4 Liquid-crystalline (LC) anthracene derivatives3–5 are promising candidates for further functionalization because controlled assembly is achieved for LC materials.6 So far, cubic,3 columnar,3 smectic4,5a–e and nematic5d,e LC anthracene derivatives have been reported. We recently prepared anthracene-based stimuli-responsive photoluminescent liquid crystals3 that change their emission colors upon switching the assembled structures via mechanical and/or thermal stimuli.
9,10-Bis(phenylethynyl)anthracene7,8 is a functional molecule that shows electro-7 and photo-functions.8 Our intention here is to develop smectic LC materials based on this moiety and to tune their photophysical properties through control of self-assembled structures.
Here we compare two 9,10-bis(phenylethynyl)anthracene derivatives 1 and 2 (Fig. 1), and report on unusual effects of tert-butyl substituents on assembly. Usually bulky substituents are used to disturb intermolecular interactions and to suppress excimer emission because of their steric effects.9 But in the present design they instead induce excimer formation.
 | ||
| Fig. 1 Molecular structures of 1 and 2. | ||
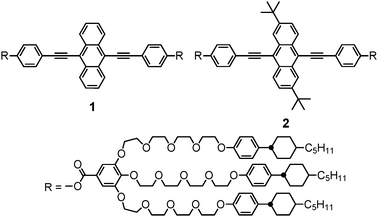
Our molecular design and the concept of this study are shown in Fig. 1 and 2. 9,10-Bis(phenylethynyl)anthracene is used as the luminescent core of compounds 1 and 2. The anthracene moiety of 1 is not substituted, while compound 2 has tert-butyl substituents on the 2- and 6-positions of the anthracene moiety.
 | ||
| Fig. 2 Molecular design for self-assembled structures of compounds 1 and 2; (a) a possible model of aggregation for 2 in the excited state. Blue and red structures indicate different molecules. Hydrogen atoms and forklike mesogens are omitted for clarity. (b) Possible structures of excimers formed for 1 and 2 in the condensed states. Mesogenic moieties of 1 and 2 are omitted in the illustration to the right of each molecular structure. | ||
A possible molecular model for 2 in the excited state is shown in Fig. 2a. Luminescent cores of 2 can approach each other up to about 3 Å and form excimers that avoid the steric hindrance of tert-butyl substituents; the relative orientation of the two molecules is partially limited by the tert-butyl substituents. On the other hand, compound 1 can form various kinds of partial-overlap excimers in addition to the normal (i.e. well-overlapped) excimer because 1 has no substituents to control the structures of excimers (Fig. 2b). Consequently, we expected that the ratio of the normal excimer is higher for 2 than 1, leading to a red-shift in excimer emission.
The luminescent core of 2 was synthesized from 2,6-di-tert-butylanthracene.13 Detailed synthetic procedures are described in the ESI.† Forklike mesogenic moieties consisting of tetraoxyethylene and p-(4-trans-pentylcyclohexyl)phenyl moieties10 were attached onto the terminal ends of each luminescent moiety to induce smectic LC properties. We previously designed and prepared the smectic liquid crystals that have chromophores4 and complex functional moieties such as rotaxane11 and catenane.11d,12 Compounds 1 and 2 are also expected to show smectic LC properties.
Compounds 1 and 2 show glass transitions at −22 °C in heating processes and smectic A (SmA) phases from 76 °C to 85 °C and 53 °C to 63 °C, respectively. The LC properties of compounds 1 and 2 are summarized in the ESI† (Table S1). Although 1 and 2 have the same luminescent cores, their photoluminescent colors in the SmA phases are significantly different (Fig. 3a). Compound 1 shows yellow-green photoluminescence, while compound 2 displays an orange emission in the SmA phases under UV irradiation (λex = 365 nm). Fig. 3b shows the emission spectra of 1 and 2 in the SmA phases. The emission spectrum of 1 in the SmA phase displays a broad emission band with an emission peak at 552 nm (Fig. 3b, black line). In contrast, 2 in the SmA phase displays a narrower emission band and the emission peak is observed at 624 nm (Fig. 3b, red line), which is a 72 nm bathochromic shift compared to that of 1. The unusual steric effect observed for 1 and 2 implies that the use of the bulky substituents can control the structures of the excimers in condensed states.
 | ||
| Fig. 3 (a) Photoluminescent images of 1 at 80 °C (top) and 2 at 55 °C (bottom) in the smectic A phases. The photoluminescent images were taken for 1 and 2 between quartz plates under UV irradiation at 365 nm. (b) Emission spectra of 1 at 80 °C (black) and 2 at 55 °C (red) in the smectic A phase (λex = 420 nm). | ||
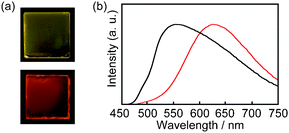
The polarizing optical micrograph of 1 taken at 80 °C upon cooling from the isotropic phase shows the focal conic textures typical of the SmA phase (Fig. 4a). Some part shows a dark image because of the homeotropic alignment of 1 in the SmA phase. The X-ray diffraction (XRD) pattern of 1 at 80 °C (Fig. 4b) indicates that compound 1 forms interdigitated layer structures (Fig. 5). Some of the terminal mesogens may bend into the anthracene part. The formation of the interdigitated layer structures in the SmA phase has been reported for several dumbbell-shaped molecules.4,14 Compound 2 also shows the interdigitated SmA phase (see, Fig. S1, ESI†). In the case of 2, bulky luminescent cores may disturb the LC structure, leading to a lower isotropization temperature than that of 1. Upon further cooling, compounds 1 and 2 exhibit monolayer smectic phases. The phase transition properties of 1 and 2 are similar to those of our previously reported compound with a 2,6-bis(phenylethynyl)anthracene moiety.4a
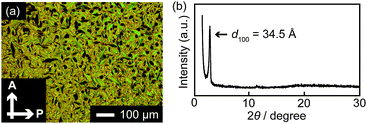 | ||
| Fig. 4 (a) Polarizing optical micrograph of 1 at 80 °C and (b) XRD pattern of 1 at 80 °C. | ||
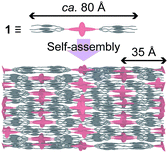 | ||
| Fig. 5 A schematic illustration of the self-assembled structure for 1 in the smectic A phase. | ||
Spectroscopic measurements of 1 and 2 were performed to obtain insight into the differences in their luminescence in the SmA phases (Fig. 3). The absorption spectra of 1 and 2 in chloroform solution (1.0 × 10−5 M) show bands between 350 nm and 500 nm (S0–S1 transition) with vibronic structures, indicative of the monomeric states of 1 and 2 (Fig. 6a). The emission spectra of 1 and 2 in a chloroform solution (1.0 × 10−5 M) also show vibronic structures. Emission lifetimes of 1 and 2 measured in a dichloromethane solution are 2.6 ns and 2.9 ns, respectively (see, Fig. S2a and b, ESI†). These photophysical properties are indicative of the monomeric states of 1 and 2. In addition, the absorption and emission properties of 1 and 2 are almost the same. It is concluded that the 2,6-tert-butyl substitutions affect little the photophysical properties of 9,10-bis(phenylethynyl)anthracene in the monomeric state. The absorption spectra of 1 and 2 in the SmA phases also show similar absorption bands to those in the chloroform solution (Fig. 6b), indicating that there is no obvious ground state interaction between the luminescent cores of 1 and 2 in the SmA phases.
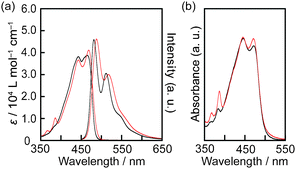 | ||
| Fig. 6 (a) Absorption and emission spectra of 1 (black) and 2 (red) in chloroform solution (1.0 × 10−5 M) at room temperature (λex = 440 nm). (b) Absorption spectra of 1 at 80 °C (black) and 2 at 55 °C (red) in the SmA phases. | ||
Although the photophysical properties in solution and the absorption properties in the SmA phases are similar for 1 and 2, the emission spectra of 1 and 2 in the SmA phases are significantly different as described above (Fig. 3b). The broad emission band of 1 and 2 observed in the emission spectra indicates that the photoluminescence of 1 and 2 in the SmA phases is attributed to the excimer formation of the 9,10-bis(phenylethynyl)anthracene moieties and that the difference in the structures of the excimers may result in the difference in the emission properties. Some 9,10-bis(phenylethynyl)anthracene derivatives were reported to exhibit excimer emission3b,5e,8 and our previously reported compound3b shows partial-overlap excimers in addition to normal excimers. The partial-overlap excimers display emission bands at shorter wavelengths than those observed for normal excimers.
The emission lifetime measurements of 1 and 2 support the differences in excimer formation (see Fig. S2c and d, ESI†). A longer emission lifetime component of 8.1 ns is observed for 1 in the SmA phase (see Fig. S2c, ESI†) than in dichloromethane solution (2.6 ns). For compound 2, a longer lifetime component of 33 ns is observed in the SmA phase (see Fig. S2d, ESI†). Our previous study3b on the relationship between lifetime and the structures of excimers for a 9,10-bis(phenylethynyl)anthracene derivative in condensed states suggests that the emission from partial excimers is dominant for 1 and compound 2 shows the emission from the normal excimer in the SmA phases.
A possible reason for the bathochromic shift in the emission of 2 is described below. In the ground state, there may be no intermolecular interaction among adjacent molecules of 1 and 2 in the SmA phases but some of the luminescent moieties form excimers after excitation. In the case of 2, the normal excimer is mainly formed because of the steric effects. The monomeric emission is little observed because energy transfer may occur from most of the excited luminescent moieties to the excimer sites. On the other hand, compound 1 without tert-butyl substituent can form various kinds of partial-overlap excimers as well as the normal excimer and the ratio of the normal excimer is low, resulting in the broader and blue-shifted emission compared to compound 2.
In summary, luminescent smectic LC materials 1 and 2 based on a 9,10-bis(phenylethynyl)anthracene moiety have been prepared and they show smectic A phases. Significant differences in photophysical properties between 1 and 2 are observed in the smectic A phase. Bulky tert-butyl substituents enhance the possibility to form well-overlapped excimers. This approach may provide a new tool to control the photoluminescent properties of π-conjugated molecular assemblies in addition to hydrogen bonding, charge transfer interactions and other interactions and may lead to the further functionalization of organic materials.
We thank Prof. K. Araki and Dr T. Mutai for their help with the measurements of emission lifetime. This work was supported by a Grant-in-Aid for Scientific Research (No. 22107003) in the Innovative Area of “Fusion Materials” (Area no. 2206) and a Grant-in-Aid for The Global COE Program “Chemistry Innovation through Cooperation of Science and Engineering” from the Ministry of Education, Culture, Sports, Science and Technology (MEXT). S.Y. and Y.S. are grateful for financial support from the Japan Society for the Promotion of Science (JSPS) Research Fellowship for Young Scientists. We thank Prof. C. L. Fraser and Prof. M. Hasegawa for fruitful discussion.
Notes and references
- (a) F. Silvestri, A. Marrocchi, M. Seri, C. Kim, T. J. Marks, A. Facchetti and A. Taticchi, J. Am. Chem. Soc., 2010, 132, 6108 CrossRef CAS; (b) H. Meng, F. Sun, M. B. Goldfinger, G. D. Jaycox, Z. Li, W. J. Marshall and G. S. Blackman, J. Am. Chem. Soc., 2005, 127, 2406 CrossRef CAS; (c) K. Ito, T. Suzuki, Y. Sakamoto, D. Kubota, Y. Inoue, F. Sato and S. Tokito, Angew. Chem., Int. Ed., 2003, 42, 1159 CrossRef CAS.
- (a) T. Hinoue, M. Miyata, I. Hisaki and N. Tohnai, Angew. Chem., Int. Ed., 2012, 51, 155 CrossRef CAS; (b) Y. Mizobe, N. Tohnai, M. Miyata and Y. Hasegawa, Chem. Commun., 2005, 1839 RSC; (c) J. Shi and C. W. Tang, Appl. Phys. Lett., 2002, 80, 3201 CrossRef CAS.
- (a) Y. Sagara, S. Yamane, T. Mutai, K. Araki and T. Kato, Adv. Funct. Mater., 2009, 19, 1869 CrossRef CAS; (b) Y. Sagara and T. Kato, Angew. Chem., Int. Ed., 2011, 50, 9128 CrossRef CAS.
- (a) S. Yamane, Y. Sagara and T. Kato, Chem. Commun., 2009, 3597 RSC; (b) S. Yamane, Y. Sagara, T. Mutai, K. Araki and T. Kato, J. Mater. Chem. C 10.1039/C3TC00861D.
- (a) S. Norvez, F.-G. Tournilhac, P. Bassoul and P. Herson, Chem. Mater., 2001, 13, 2552 CrossRef CAS; (b) S. Méry, D. Haristoy, J.-F. Nicoud, D. Guillon, H. Monobe and Y. Shimizu, J. Mater. Chem., 2003, 13, 1622 RSC; (c) Y. Takaguchi, T. Tajima, Y. Yanagimoto, S. Tsuboi, K. Ohta, J. Motoyoshiya and H. Aoyama, Org. Lett., 2003, 5, 1677 CrossRef CAS; (d) A. N. Cammidge and V. H. M. Goddard, Liq. Cryst., 2008, 35, 1145 CrossRef CAS; (e) R. Giménez, M. Piñol and J. L. Serrano, Chem. Mater., 2004, 16, 1377 CrossRef.
- (a) Handbook of Liquid Crystals, ed. D. Demus, J. W. Goodby, G. W. Gray, H.-W. Spiess and V. Vill, Wiley-VCH, Weinheim, 1998 Search PubMed; (b) T. Kato, Science, 2002, 295, 2414 CrossRef CAS; (c) D. Guillon and R. Deschenaux, Curr. Opin. Solid State Mater. Sci., 2002, 6, 515 CrossRef CAS; (d) G. H. Mehl, Angew. Chem., Int. Ed., 2005, 44, 672 CrossRef CAS; (e) T. Kato, N. Mizoshita and K. Kishimoto, Angew. Chem., Int. Ed., 2006, 45, 38 CrossRef CAS; (f) C. Damm, G. Israel, T. Hegmann and C. Tschierske, J. Mater. Chem., 2006, 16, 1808 RSC; (g) C. Tschierske, Chem. Soc. Rev., 2007, 36, 1930 RSC; (h) I. M. Saez and J. W. Goodby, Struct. Bonding, 2008, 128, 1 CrossRef CAS; (i) T. Kato, T. Yasuda, Y. Kamikawa and M. Yoshio, Chem. Commun., 2009, 729 RSC; (j) S. Frein, F. Camerel, R. Ziessel, J. Barberá and R. Deschenaux, Chem. Mater., 2009, 21, 3950 CrossRef CAS; (k) S.-J. Yoon, J. H. Kim, K. S. Kim, J. W. Chung, B. Heinrich, F. Mathevet, P. Kim, B. Donnio, A.-J. Attias, D. Kim and S. Y. Park, Adv. Funct. Mater., 2012, 22, 61 CrossRef CAS; (l) A. Yoshizawa, Polym. J., 2012, 44, 490 CrossRef CAS; (m) B. R. Wiesenauer and D. L. Gin, Polym. J., 2012, 44, 461 CrossRef CAS.
- (a) C. Wang, Y. Liu, Z. Ji, E. Wang, R. Li, H. Jiang, Q. Tang, H. Li and W. Hu, Chem. Mater., 2009, 21, 2840 CrossRef CAS; (b) L. Valentini, D. Bagnis, A. Marrocchi, M. Seri, A. Taticci and J. M. Kenny, Chem. Mater., 2008, 20, 32 CrossRef CAS.
- (a) M. Levitus and M. A. Garcia-Garibay, J. Phys. Chem. A, 2000, 104, 8632 CrossRef CAS; (b) A. D. Malakhov, M. V. Skorobogatyi, I. A. Prokhorenko, S. V. Gontarev, D. T. Kozhich, D. A. Stetsenko, I. A. Stepanova, Z. O. Shenkarev, Y. A. Berlin and V. A. Korshun, Eur. J. Org. Chem., 2004, 1298 CrossRef CAS.
- (a) D. Thirion, C. Poriel, R. Métivier, J. Rault-Berthelot, F. Barrière and O. Jeannin, Chem.–Eur. J., 2011, 17, 10272 CrossRef CAS; (b) A. C. Benniston, A. Harriman, S. L. Howell, C. A. Sams and Y.-G. Zhi, Chem.–Eur. J., 2007, 13, 4665 CrossRef CAS.
- Y. Iinuma, K. Kishimoto, Y. Sagara, M. Yoshio, T. Mukai, I. Kobayashi, H. Ohno and T. Kato, Macromolecules, 2007, 40, 4874 CrossRef CAS.
- (a) I. Aprahamian, T. Yasuda, T. Ikeda, S. Saha, W. R. Dichtel, K. Isoda, T. Kato and J. F. Stoddart, Angew. Chem., Int. Ed., 2007, 46, 4675 CrossRef CAS; (b) I. Aprahamian, O. Š. Miljanić, W. R. Dichtel, K. Isoda, T. Yasuda, T. Kato and J. F. Stoddart, Bull. Chem. Soc. Jpn., 2007, 80, 1856 CrossRef CAS; (c) T. Yasuda, K. Tanabe, T. Tsuji, K. K. Coti, I. Aprahamian, J. F. Stoddart and T. Kato, Chem. Commun., 2010, 46, 1224 RSC; (d) J. Sakuda, T. Yasuda and T. Kato, Isr. J. Chem., 2012, 52, 854 CrossRef CAS.
- (a) E. D. Baranoff, J. Voignier, T. Yasuda, V. Heitz, J.-P. Sauvage and T. Kato, Angew. Chem., Int. Ed., 2007, 46, 4680 CrossRef CAS; (b) E. D. Baranoff, J. Voignier, T. Yasuda, V. Heitz, J.-P. Sauvage and T. Kato, Mol. Cryst. Liq. Cryst., 2009, 509, 165 CrossRef.
- (a) P. P. Fu and R. G. Harvey, J. Org. Chem., 1977, 42, 2407 CrossRef CAS; (b) K. Danel and J. T. Lin, ARKIVOC, 2002, 12 CAS; (c) J.-F. Lee, Y.-C. Chen, J.-T. Lin, C.-C. Wu, C.-Y. Chen, C.-A. Dai, C.-Y. Chao, H.-L. Chen and W.-B. Liau, Tetrahedron, 2011, 67, 1696 CrossRef CAS.
- A. Yamaguchi, N. Uehara, J. Yamamoto and A. Yoshizawa, Chem. Mater., 2007, 19, 6445 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, liquid-crystalline properties and time-resolved fluorescence decay profiles for 1 and 2. See DOI: 10.1039/c3cc00072a |
| This journal is © The Royal Society of Chemistry 2013 |