 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unbridged Au(II)–Au(II) bonds are theoretically allowed†
Xiao-Gen
Xiong
ab and
Pekka
Pyykkö
*b
aDepartment of Chemistry, Tsinghua University, 100 084 Beijing, China
bDepartment of Chemistry, University of Helsinki, P.O.B. 55, FI-00014 Helsinki, Finland. E-mail: Pekka.Pyykko@helsinki.fi; Fax: +358 9 191 50169; Tel: +358 9 191 50171
First published on 4th January 2013
Abstract
The bonding in the unbridged closed-shell Au(II)–Au(II) dimers X4Au2(C5H5N)2, X = H, F–I and CF3, is analyzed and the short Au–Au bonds around 250 pm are reproduced by a novel 6s6pz5dxy hybridization.
Short Au(II)–Au(II) bonds of 269.7(1)–297.7(10) pm are experimentally known in systems like [Au2(i-MNT)2]2− (i-MNT:1,1-dicyanoethene-2,2-dithiolate).1 Common to all of them is that the two ligand atoms are coupled by a bridge. The bonding in these systems has been analyzed using semiempirical methods by Hoffmann's group2 and later, using ab initio methods, by us.3 Recently two cases with unbridged Au(II)–Au(II) bonds were reported.4,5 Earlier examples were reported by the groups of Yam6,7 and Raubenheimer.8 Common to all these cases were nearly perpendicular Au (CN = 4) planes and short Au(II)–Au(II) bonds of the order of 249–264 pm. We here account for both observations by performing a DFT study on the model systems X4Au2(C5H5N)2, X = H, F–I and CF3. It should be noted that, in contrast to the ubiquitous Au(I)⋯Au(I) closed-shell “aurophilic” attractions which are basically of dispersion type,1 we here expect a dominant covalent bond.
The optimized PBE geometries of the model systems9 are listed in Table 1. All of them have a very short unsupported Au(II)–Au(II) bond. One thing that should be noted is that, in all cases, the N–Au–Au–N backbones are linear. All molecules except (CF3)4Au2(C5H5N)2 are confirmed to possess D2 symmetry, while the latter one has C2 symmetry. The R(Au–Au) of (CF3)4Au2(C5H5N)2 in our calculation is 255.3 pm, while one experimental value is 250.62(9) pm.4 Note the position of –CF3 between F and Cl, coherent with the electronegativity data of García et al.10 Further calculations with hybrid, meta-GGA and hybrid meta-GGA functionals were also carried out; the geometries are listed in the ESI† and the results also confirm the existence of a short, unsupported Au(II)–Au(II) bond.
| X | R(Au–Au) | R(Au–N) | R(Au–X) | ∠XAuX | ∠XAuN |
|---|---|---|---|---|---|
| H | 250.7 | 217.0 | 165.4 | 173.5 | 93.3 |
| F | 254.4 | 218.1 | 196.9 | 179.0 | 89.5 |
| Cl | 256.6 | 220.4 | 231.9 | 174.7 | 92.7 |
| Br | 256.5 | 219.0 | 248.3 | 174.0 | 93.0 |
| I | 257.4 | 219.0 | 267.0 | 173.0 | 93.5 |
| CF3 | 255.3 | 217.5 | 212.2 | 178.2 | 89.1 |
In order to investigate the Au(II)–Au(II) bond, we carried out a population analysis and calculated bond orders using a variety of theoretical approaches.11–18 The calculated atomic charges and bond orders are given in Tables 2 and 3. All methods show that the two Au atoms are positively charged. The calculated electron localization functions (ELFs, in Fig. 1) show that there is clearly electron pair density between the two Au atoms. The Au(II)–Au(II) bonding energy curves are shown in Fig. 2. Here the bonding energy is defined as the energy difference between the complex and two X2Au(C5H5N) monomers. The geometries of two monomers are fixed to the optimized X4Au2(C5H5N)2 geometry and only the distance of two Au atoms is varied. The Au–Au bonding energy is approximately 200 kJ mol−1, and varied with the ligands. The bonding energies at the equilibrium geometry are listed in Table 4.
| X | Atom | Hirshfeld | Voronoi | MDC-q | AIM |
|---|---|---|---|---|---|
| H | Au | 0.12 | 0.29 | 0.56 | 0.25 |
| H | −0.17 | −0.26 | −0.37 | −0.22 | |
| N | −0.06 | −0.07 | 0.42 | −0.96 | |
| F | Au | 0.37 | 0.36 | 0.74 | 0.92 |
| F | −0.31 | −0.32 | −0.52 | −0.60 | |
| N | −0.06 | −0.06 | −0.32 | −0.97 | |
| Cl | Au | 0.29 | 0.31 | 0.35 | 0.59 |
| Cl | −0.25 | −0.28 | −0.33 | −0.42 | |
| N | −0.06 | −0.07 | 0.01 | −0.95 | |
| Br | Au | 0.25 | 0.26 | 0.43 | 0.45 |
| Br | −0.23 | −0.26 | −0.37 | −0.34 | |
| N | −0.06 | −0.07 | 0.19 | −1.01 | |
| I | Au | 0.20 | 0.19 | 0.27 | 0.26 |
| I | −0.21 | −0.22 | −0.28 | −0.25 | |
| N | −0.07 | −0.07 | 0.33 | −1.02 | |
| CF3 | Au | 0.25 | 0.25 | 0.41 | 0.48 |
| C | 0.10 | 0.08 | −0.09 | 1.44 | |
| N | −0.06 | −0.08 | −0.34 | −1.00 | |
| X | Bond | Mayer | G–J | N–M(3) |
|---|---|---|---|---|
| H | Au–Au | 0.77 | 0.46 | 0.27 |
| Au–N | 0.40 | 0.24 | 0.21 | |
| Au–H | 0.76 | 0.43 | 0.46 | |
| F | Au–Au | 0.74 | 0.43 | 0.22 |
| Au–N | 0.38 | 0.24 | 0.20 | |
| Au–F | 0.59 | 0.47 | 0.74 | |
| Cl | Au–Au | 0.67 | 0.38 | 0.20 |
| Au–N | 0.40 | 0.25 | 0.21 | |
| Au–Cl | 0.78 | 0.49 | 0.65 | |
| Br | Au–Au | 0.66 | 0.38 | 0.20 |
| Au–N | 0.41 | 0.25 | 0.21 | |
| Au–Br | 0.67 | 0.47 | 0.61 | |
| I | Au–Au | 0.65 | 0.36 | 0.21 |
| Au–N | 0.42 | 0.25 | 0.22 | |
| Au–I | 0.79 | 0.46 | 0.56 | |
| CF3 | Au–Au | 0.83 | 0.42 | 0.22 |
| Au–N | 0.44 | 0.14 | 0.19 | |
| Au–C | 0.74 | 0.45 | 0.33 | |
 | ||
| Fig. 1 The electron localization functions (ELFs) of (CF3)4Au2(C5H5N)2. From left, the atoms are N–Au–Au–N. | ||
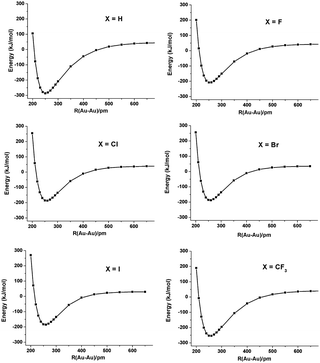 | ||
| Fig. 2 Au(II)–Au(II) bonding energy curves of X4Au2(C5H5N)2, X = H, F–I and CF3. The geometry of X2Au(C5H5N) was fixed to the monomer geometry in the complex. | ||
| X | H | F | Cl | Br | I | CF3 |
|---|---|---|---|---|---|---|
| Bonding energy | 286.3 | 209.1 | 188.1 | 188.5 | 185.7 | 255.2 |
The Kohn–Sham bonding orbitals for the Au–Au bonds are of the, perhaps less obvious, types given in Fig. 3. Here the X = H is selected. The experimental case X = –CF3 is shown in the graphical abstract. Both cases show an spd or more precisely 6s5dxy6pz hybridization. The percentages for X = –CF3 are 6s 31%, 5d 12%, 6pz 10%. We have not previously seen a hybridization like this.19–21
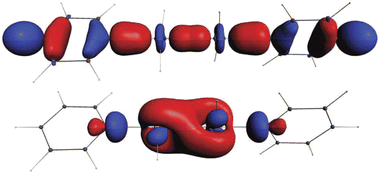 | ||
| Fig. 3 The Au(II)–Au(II) bonding orbitals of H4Au2(C5H5N)2 (isosurf. = 0.05 a.u.). For the real-world case X = –CF3, see the graphical abstract. | ||
In order to probe the stabilizing effect of the axial ligand, two other ligands, –CH3 and NH3, were chosen to replace pyridine. The unbridged Au(II)–Au(II) bond also exists when pyridine is replaced by NH3, but –CH3, leading to Au(III), will destabilize the Au–Au bond, see the ESI.†
In conclusion, using DFT calculations, we investigated the unsupported Au(II)–Au(II) bond in the model system X4Au2(C5H5N)2, X = H, F–I and CF3. The bond length of Au–Au is located in the covalent Au–Au region. All the theoretical results clearly show the covalent nature of the unsupported Au(II)–Au(II) bond in these molecules.
Computational methods: all calculations were performed with density functional theory (DFT). Generalized gradient approximation (GGA) with the PBE exchange–correlation functional9 implemented in the Amsterdam Density Functional program (ADF 2010.01)22 was used. The Slater basis sets with the quality of triple-ζ plus two polarization functions (TZ2P) with the frozen-core approximation applied to inner shells were used. The scalar-relativistic (SR) effects were taken into account by the zero-order-regular approximation (ZORA).23 Geometries were fully optimized at the SR-ZORA level. Vibrational frequency calculations were also carried out at the SR-ZORA level with the PBE functional to confirm the minima. In order to confirm our results, we performed further DFT calculations with hybrid, meta-GGA and hybrid meta-GGA functionals, implemented in Gaussian09.24 The computational details of Gaussian calculations can be found in the ESI.† Spin–orbit effects were tested at the ZORA level in the X = H case and found negligible.
The work of XGX at Helsinki is supported by the China Scholarship Council. Computer resources were obtained from the Centre for Scientific Computing, Finland.
References
- P. Pyykkö, Chem. Rev., 1997, 97, 597–636 CrossRef.
- Y. Jiang, S. Alvarez and R. Hoffmann, Inorg. Chem., 1985, 24, 749–757 CrossRef CAS.
- P. Pyykkö and F. Mendizabal, Inorg. Chem., 1998, 37, 3018–3025 CrossRef.
- D. Zopes, C. Hegemann, W. Tyrra and S. Mathur, Chem. Commun., 2012, 48, 8805–8807 RSC.
- D.-A. Roşca, D. A. Smith, D. L. Hughes and M. Bochmann, Angew. Chem., Int. Ed., 2012, 51, 10643–10646 Search PubMed.
- V. W.-W. Yam, S. W.-K. Choi and K.-K. Cheung, Chem. Commun., 1996, 1173–1174 RSC.
- V. W.-W. Yam, C.-K. Li, C.-L. Chan and K.-K. Cheung, Inorg. Chem., 2001, 40, 7054–7058 CrossRef CAS.
- J. Coetzee, W. F. Gabrielli, K. Coetzee, O. Schuster, S. D. Nogai, S. Cronje and H. G. Raubenheimer, Angew. Chem., Int. Ed., 2007, 46, 2497–2500 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- A. García-Monforte, S. Martínez-Salvador and B. Menjón, Eur. J. Inorg. Chem., 2012, 4945–4966 Search PubMed.
- F. L. Hirshfeld, Theor. Chem. Acc., 1977, 44, 129–138 CAS.
- F. M. Bickelhaupt, N. J. R. van Eikema Hommes, C. Fonseca Guerra and E. J. Baerends, Organometallics, 1996, 15, 2923–2931 CrossRef CAS.
- M. Swart, P. T. van Duijnen and J. G. Snijders, J. Comput. Chem., 2001, 22, 79–88 CrossRef CAS.
- J. I. Rodríguez, R. F. Bader, P. W. Ayers, C. Michel, A. W. Götz and C. Bo, Chem. Phys. Lett., 2009, 472, 149–152 CrossRef CAS.
- J. I. Rodríguez, A. M. Köster, P. W. Ayers, A. Santos-Valle, A. Vela and G. Merino, J. Comput. Chem., 2009, 30, 1082–1092 CrossRef CAS.
- I. Mayer, Chem. Phys. Lett., 1983, 97, 270–274 CrossRef CAS.
- M. S. Gopinathan and K. Jug, Theor. Chem. Acc., 1983, 63, 497–509 Search PubMed.
- A. Michalak, R. L. DeKock and T. Ziegler, J. Phys. Chem. A, 2008, 112, 7256–7263 CrossRef CAS.
- Note that the bonding orbital in Fig. 3b has no conspicuous nodes in the Au–Au part while a dxy–dxy bond would have two. Such a “screw, don't split” principle is not unlike the “angular nodes” explanation for the 18e principle.
- P. Pyykkö, J. Organomet. Chem., 2006, 691, 4336–4340 CrossRef.
- The dxy on atom A bonds here to the dx2−y2 on atom B.
- http://www.scm.com .
- E. van Lenthe, R. van Leeuwen, E. J. Baerends and J. G. Snijders, Int. J. Quantum Chem., 1996, 57, 281–293 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision A.1, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cc37875b |
| This journal is © The Royal Society of Chemistry 2013 |