Facile carbon–sulfur bond cleavage in diarylsulfonium ylides: a catalytic sulfur-to-silicon group transfer†‡
Xueliang
Huang
,
Richard
Goddard
and
Nuno
Maulide
*
Max-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470 Mülheim an der Ruhr, Germany. E-mail: maulide@mpi-muelheim.mpg.de; Tel: +49-208-306-2450
First published on 11th September 2012
Abstract
An unusual cleavage of the non-ylidic carbon–sulfur bond of a sulfonium ylide is reported. The reaction can be catalysed by a variety of palladium(II) complexes under mild conditions. Preliminary results show that coordination of the ylide to the metal center results in significant structural changes.
Sulfonium ylides are classical textbook reagents for three-membered ring formation reactions,1 and have recently been the subject of renewed attention as potential carbene donors to a metal promoter.2 Such carbene transfers are thought to occur either directly, by release of the corresponding neutral sulfide upon interaction with a metal catalyst, or by a “rebound” type mechanism of nucleophilic attack to an activated carbon center followed by leaving group expulsion (Scheme 1a).3 In contrast, to the best of our knowledge, there is no report on the catalytic activation of the non-ylidic carbon–sulfur bond of a sulfonium ylide (Scheme 1b).4 Herein, we describe a facile catalytic carbon–sulfur bond cleavage of diarylsulfonium ylides, which we believe constitutes an unprecedented reactivity mode of these reagents.
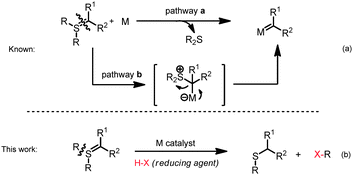 | ||
| Scheme 1 Different reactivities of sulfonium ylides. | ||
We recently reported a simple synthesis of diphenyl sulfonium ylides by a new concept of “ylide transfer”.5 While investigating the reactivity towards metal catalysts of some of the products that can be obtained by that methodology,6 we have discovered that an unusual cleavage of the sulfur–aryl bond takes place through palladium catalysis (Scheme 2). In the event, exposure of indole-derived diphenylsulfonium ylide 1a to 20 mol% of palladium(II) acetate in the presence of phenyldimethylsilane as a reducing agent generated 3-(phenylsulfanyl)indole 3a in 57% yield along with diphenyldimethylsilane (as detected by GC-MS).7,8
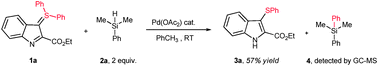 | ||
| Scheme 2 Discovery of a catalytic aryl–sulfur bond cleavage and transfer. | ||
Subsequent optimisation studies revealed the necessity of a palladium(II) salt as a catalyst and the superior performance of silanes as reducing agents. As depicted in Table 1, the reaction can take place in various solvents. In particular, clean reactions proceeding at reasonable rates were observed when acetic acid was used as the reaction medium (entry 4). In contrast, changing the solvent to ethyl acetate resulted in fast formation of sulfide 3a together with other unidentified products (entry 8). The reaction can be catalysed by diverse palladium(II) sources (entries 9, 10, 11 and 13), though complexes bearing bidentate ligands resulted in lower efficiency (entry 12). We eventually found that the use of a cationic palladium(II) source (the complex (CH3CN)4Pd(BF4)2) in the presence of acetic acid led to very high reproducible yields of the dearylated product 3a in a short reaction time (entries 14 and 15).

| Entry | Solvent | Catalyst | x | Time/h | Yielda (%) |
|---|---|---|---|---|---|
| a NMR yields determined using CH2Br2 as the internal standard. b Isolated yield. c 10 equiv. of acetic acid were added. d The reaction was carried out under air. | |||||
| 1 | PhMe | Pd(OAc)2 | 20 | 20 | 57 |
| 2 | Acetone | Pd(OAc)2 | 20 | 36 | 27 |
| 3 | MeOH | Pd(OAc)2 | 20 | 24 | 30 |
| 4 | AcOH | Pd(OAc)2 | 20 | 24 | 70b |
| 5 | DMF | Pd(OAc)2 | 20 | 14 | 56 |
| 6 | PhH | Pd(OAc)2 | 20 | 14 | 62 |
| 7 | DCE | Pd(OAc)2 | 20 | 12 | 72 |
| 8 | AcOEt | Pd(OAc)2 | 20 | 1 | 68 |
| 9 | AcOH | Pd(PhCN)2Cl2 | 20 | 24 | 64 |
| 10 | AcOH | (Ph3P)2PdCl2 | 20 | 24 | 67 |
| 11 | AcOH | (CH3CN)4Pd(BF4)2 | 20 | 24 | 74 |
| 12 | AcOH | dpppPdCl2 | 20 | 24 | 31 |
| 13 | AcOH | Pd(CH3CN)2Cl2 | 20 | 13 | 83 |
| 14c | AcOEt | (CH3CN)4Pd(BF4)2 | 20 | 1 | 81b |
| 15c | AcOEt | (CH3CN)4Pd(BF4)2 | 10 | 1 | 81b |
| 16c,d | AcOEt | (CH3CN)4Pd(BF4)2 | 5 | 1 | 91b |
We therefore took the optimised conditions for further studies (Table 1, entry 15). As portrayed in Scheme 3, this dearylation has a broad scope among most of the indole and pyrrole ylides examined,9 and tolerates electron-rich and electron-poor substituents (cf.3a to 3h). Interestingly, this palladium-catalysed reaction leaves an aromatic bromide untouched (cf.3g).
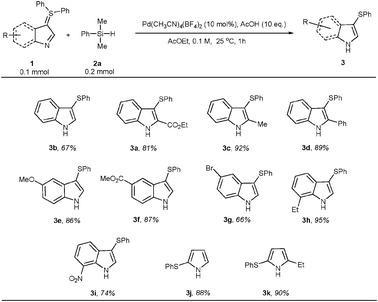 | ||
| Scheme 3 Scope of the palladium-catalysed dearylation of diphenyl sulfonium ylides. | ||
It is also possible to effect dearylation of carbonyl-substituted ylides, leading formally to the products of α-sulfenylation of the parent active methylene compounds (Scheme 4).10
 | ||
| Scheme 4 Palladium-catalysed dearylation of carbonyl-substituted diphenylsulfonium ylides. | ||
From the outset, we were intrigued by the mechanistic details of this transformation. The use of deuteriosilane as a reducing agent did not lead to noticeable deuterium incorporation in the final product (Scheme 5a). In contrast, the use of normal hydrosilane with added deuterated acetic acid provided 63% deuterium incorporation at the indole nitrogen (Scheme 5b).
 | ||
| Scheme 5 Isotope labelling experiments. | ||
It was also established through a series of background experiments that the reaction does not proceed in the absence of either the metal catalyst or the silane 2a. Interestingly, other hydride donors are also viable for this transformation. When 2 equivalents of pinacolborane 2b were employed (Scheme 6a), the sulfenyl indole 3a was obtained in good yield. Even formic acid 2c11can be used, although this requires a longer time (Scheme 6b).
 | ||
| Scheme 6 Studies on alternative reducing reagents, aPhenylboronic acid pinacol ester can be detected by GC-MS through comparison with an authentic sample. | ||
In addition, mixing sulfonium ylide 1a with Pd(OAc)2 and LiCl led to the isolation of a stable 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex as shown in Scheme 7, in which a Pd(II) centre is coordinated to the imino indole fragment of the sulfonium ylide (in its neutral form).‡ There are important geometrical differences between the structure of sulfonium ylide 1a when it acts as a ligand bound to Pd in 7 and that of the isolated sulfonium ylide in the crystal of 1a (see the ESI‡ for a tabulated comparison).5 Although the standard uncertainties of the distances are somewhat high, a distinctive trend is observable. In 7, the N
:1 complex as shown in Scheme 7, in which a Pd(II) centre is coordinated to the imino indole fragment of the sulfonium ylide (in its neutral form).‡ There are important geometrical differences between the structure of sulfonium ylide 1a when it acts as a ligand bound to Pd in 7 and that of the isolated sulfonium ylide in the crystal of 1a (see the ESI‡ for a tabulated comparison).5 Although the standard uncertainties of the distances are somewhat high, a distinctive trend is observable. In 7, the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C1 bond of the indole derived diphenylsulphonium ylide is longer, the C1–C2 bond shorter, and the C2S and S–C6 bonds are all longer than in 1a (Scheme 8). This is consistent with a loss of ylide character in the C2S bond, a build-up of electron density on the S atom and a weakening of the S–Ph bonds upon coordination of 1a to the metal. Similarly, coordination of 1a to the metal causes the C4–N–C1 and C1–C2–C3 angles in the five-membered heterocyclic ring to widen and the N–C1–C2 angle to decrease, supporting this view. The relatively short intramolecular Pd⋯O distances of 2.960(4) and 2.996(4) Å to the carbonyl groups (O1 and O3) indicate that net donation of electron density from the metal to the sulfonium ylide ligand leaves the Pd atom positively charged.
C1 bond of the indole derived diphenylsulphonium ylide is longer, the C1–C2 bond shorter, and the C2S and S–C6 bonds are all longer than in 1a (Scheme 8). This is consistent with a loss of ylide character in the C2S bond, a build-up of electron density on the S atom and a weakening of the S–Ph bonds upon coordination of 1a to the metal. Similarly, coordination of 1a to the metal causes the C4–N–C1 and C1–C2–C3 angles in the five-membered heterocyclic ring to widen and the N–C1–C2 angle to decrease, supporting this view. The relatively short intramolecular Pd⋯O distances of 2.960(4) and 2.996(4) Å to the carbonyl groups (O1 and O3) indicate that net donation of electron density from the metal to the sulfonium ylide ligand leaves the Pd atom positively charged.
 | ||
| Scheme 7 Preparation of complex 7. | ||
 | ||
| Scheme 8 Catalytic activity of complex 7. | ||
This complex is catalytically active, as demonstrated by its ability to promote dearylation of 10-fold amounts of either 1a (Scheme 8a) or 5a (Scheme 8b). From a mechanistic point of view, it is possible that the aforementioned electronic effects upon coordination favor direct attack of the silane at sulfur, followed by bond reorganisation to release the product and regenerate the palladium catalyst.
In summary, we describe an unprecedented palladium-catalysed C(aryl)–S bond cleavage of diaryl sulfonium ylides that proceeds under mild conditions with transfer of the aryl moiety from sulfur to an acceptor silane. This intriguing process, which relies on the use of palladium(II) salts, leads to the products of formal sulfenylation. Alternative acceptors such as boranes or even formic acid can be employed. Mechanistic experiments suggest that the ylide substrate can serve as a suitable neutral, 2-electron ligand for palladium(II). Further explorations of this and other unique modes of reactivity of diphenyl sulfonium ylides are underway.
Notes and references
- (a) B. M. Trost and L. S. Melvin, Sulfur Ylides;Academic Press, New York, 1975 Search PubMed; (b) A.-H. Li, L.-X. Dai and V. K. Aggarwal, Chem. Rev., 1997, 97, 2341 CrossRef CAS; (c) V. K. Aggarwal and C. L. Winn, Acc. Chem. Res., 2004, 37, 611 CrossRef CAS; (d) Y. Tang, S. Ye and X.-L. Sun, Synlett, 2005, 2720 CrossRef CAS; (e) E. M. McGarrigle, E. L. Meyers, O. Illa, M. A. Shaw, S. L. Riches and V. K. Aggarwal, Chem. Rev., 2007, 107, 5841 CrossRef CAS.
- (a) B. M. Trost, J. Am. Chem. Soc., 1966, 88, 1587 CrossRef CAS; (b) P. Müller, D. Fernandez, P. Nury and J.-C. Rossier, Helv. Chim. Acta, 1999, 82, 935 CrossRef; (c) M. Gandelman, B. Rybtchinski, N. Ashkenazi, R. M. Gauvin and D. Milstein, J. Am. Chem. Soc., 2001, 123, 5372 CrossRef CAS; (d) M. Gandelman, K. M. Naing, B. Rybtchinski, E. Poverenov, Y. Ben-David, N. Ashkenazi, R. M. Gauvin and D. Milstein, J. Am. Chem. Soc., 2005, 127, 15265 CrossRef CAS; (e) S. A. Stoffregen, M. Heying and W. S. Jenks, J. Am. Chem. Soc., 2007, 129, 15746 CrossRef CAS.
- For a review on decomposition of iodonium- and sulfonium ylides, see: (a) P. Müller, D. Fernandez, P. Nury and J.-C. Rossier, J. Phys. Org. Chem., 1998, 11, 321 CrossRef; (b) For metal carbene formation from sulfonium ylides, see ref. 2a: ; (c) T. Cohen, G. Herman, T. M. Chapman and D. Kuhn, J. Am. Chem. Soc., 1974, 96, 5627 CrossRef CAS; (d) B. Cimetière and M. Julia, Synlett, 1991, 271 CrossRef; (e) P. Müller, D. Fernandez, P. Nury and J.-C. Rossier, Helv. Chim. Acta, 1999, 82, 935 CrossRef; (f) S. Kramer and T. Skrydstrup, Angew. Chem., Int. Ed., 2012, 51, 4681 CrossRef CAS . For metal carbenes generated from diazo compounds, see: ; (g) M. P. Doyle, Chem. Rev., 1986, 86, 919 CrossRef CAS.
- For selected examples on application of the Stevens rearrangement, see: (a) M. P. Doyle, J. H. Griffin, M. S. Chinn and D. V. Leusen, J. Org. Chem., 1984, 49, 1917 CrossRef CAS; (b) V. Nair, S. M. Nair, S. Mathai, J. Liebscher, B. Ziemer and K. Narsimulu, Tetrahedron Lett., 2004, 45, 5759 CrossRef CAS; (c) K. K. Ellis-Holder, B. P. Peppers, A. Y. Kovalevsky and S. T. Diver, Org. Lett., 2006, 8, 2511 CrossRef CAS; (d) A. J. P. Mortimer, A. E. Aliev, D. A. Tocher and M. J. Porter, Org. Lett., 2008, 10, 5477 CrossRef CAS; (e) J.-P. Qu, Z.-H. Xu, J. Zhou, C.-L. Cao, X.-L. Sun, L.-X. Dai and Y. Tang, Adv. Synth. Catal., 2009, 351, 308 CrossRef CAS.
- X. Huang, R. Goddard and N. Maulide, Angew. Chem., Int. Ed., 2010, 49, 8979 CrossRef CAS.
- (a) X. Huang, B. Peng, M. Luparia, L. F. R. Gomes, L. F. Veiros and N. Maulide, Angew. Chem., Int. Ed., 2012, 51, 8886 CrossRef CAS.
- For selected examples on sulfenylation of indoles, see: (a) M. Tudge, M. Tamiya, C. Savarin and G. R. Humphrey, Org. Lett., 2006, 8, 565 CrossRef CAS; (b) Y.-J. Guo, R.-Y. Tang, J.-H. Li, P. Zhong and X.-G. Zhang, Adv. Synth. Catal., 2009, 351, 2615 CrossRef CAS; (c) J. S. Yadav, B. V. S. Reddy, Y. J. Reddy and K. Praneeth, Synthesis, 2009, 1520 CrossRef CAS; (d) X.-L. Fang, R.-Y. Tang, P. Zhong and J.-H. Li, Synthesis, 2009, 4183 CAS; (e) Z. Li, J. Hong and X. Zhou, Tetrahedron, 2011, 67, 3690 CrossRef CAS; (f) D. Huang, J. Chen, W. Dan, J. Ding, M. Liu and H. Wu, Adv. Synth. Catal., 2012, 354, 2123 CrossRef CAS.
- B. Marciniec, Hydrosilylation, Advances in Silicon Science, Springer, 2009 Search PubMed.
- For selected examples on preparation of sulfanyl pyrroles, see: H. M. Gillis, L. Greene and A. Thompson, Synlett, 2009, 112 CAS.
- M. A. Rashid, N. Rasool, M. Adeel, H. Reinke, C. Fischer and P. Langer, Tetrahedron, 2008, 64, 3782 CrossRef CAS.
- For hydrogen transfer from formic acid catalysed by palladium, see: (a) B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 1996, 118, 6305 CrossRef CAS; (b) B. M. Trost, F. D. Toste and K. Greenman, J. Am. Chem. Soc., 2003, 125, 4518 CrossRef CAS; (c) R. Nakao, H. Rhee and Y. Uozumi, Org. Lett., 2005, 7, 163 CrossRef CAS; (d) S. K. Pandey, A. E. Greene and J.-F. Poisson, J. Org. Chem., 2007, 72, 7769 CrossRef CAS; (e) J. M. Brunel, Synlett, 2007, 330 CrossRef CAS; (f) R. Shen, T. Chen, Y. Zhao, R. Qiu, Y. Zhou, S. Yin, X. Wang, M. Goto and L.-B. Han, J. Am. Chem. Soc., 2011, 133, 17037 CrossRef CAS.
Footnotes |
| † This article is part of the ChemComm ‘Emerging Investigators 2013’ themed issue. |
| ‡ Electronic supplementary information (ESI) available: Experimental, X-ray crystallography data and NMR. CCDC 894535. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2cc35762c |
| This journal is © The Royal Society of Chemistry 2013 |