Enhanced drug delivery to hepatocellular carcinoma with a galactosylated core–shell polyphosphoester nanogel†
Juan Wu‡a,
Tian-Meng Sun‡a,
Xian-Zhu Yang*ab,
Jing Zhua,
Xiao-Jiao Dua,
Yan-Dan Yaoc,
Meng-Hua Xionga,
Hong-Xia Wanga,
Yu-Cai Wanga and
Jun Wang*a
aHefei National Laboratory for Physical Sciences at the Microscale and School of Life Sciences, University of Science and Technology of China, Hefei, Anhui 230027, People's Republic of China. E-mail: jwang699@ustc.edu.cn; Fax: +86 551 63600402; Tel: +86 551 63600335
bSchool of Medical Engineering, Hefei University of Technology, Hefei, Anhui 230027, People's Republic of China. E-mail: yangxz@hfut.edu.cn; Fax: +86 551 62905460; Tel: +86 551 62905460
cDepartment of Breast Surgery, No. 2 Affiliated Hospital of Sun Yat-Sen University, Guangzhou 510120, People's Republic of China
First published on 17th July 2013
Abstract
Effective systemic therapy is often necessary to treat hepatocellular carcinoma (HCC). We synthesized a Gal-PPE nanogel consisting of a cross-linked polyphosphate core and galactosylated poly(ethylene glycol) arms for enhanced doxorubicin delivery to diethylnitrosamine-induced HCC in rats. The Gal-PPE nanogel exhibited high affinity to HepG2 cells in vitro, mediated by the asialoglycoprotein receptor. In vivo studies revealed that the Gal-PPE nanogel was taken up more efficiently by hepatocytes, in contrast to m-PPE nanogel. Consequently, doxorubicin delivery with Gal-PPE significantly inhibited the progress of HCC, reducing neoplastic liver nodules and prolonging the survival time of HCC rats more significantly. These results demonstrate the potential of Gal-PPE as a nanocarrier for improved HCC chemotherapy.
1 Introduction
Primary liver cancer is a major health problem worldwide, and hepatocellular carcinoma (HCC) is the most common primary cancer of the liver. Surgical resection is the mainstay of treatment for HCC.1 However, it is very limited for patients with multiple or metastatic tumors. Effective chemotherapeutic agents to improve the survival rate of patients are desired.Nanoparticles as vehicles for drug molecules for diagnostics and therapeutics have been one of the major focuses in nanomedicine.2,3 Recent studies have successfully demonstrated that with optimization certain nanoparticles can effectively deliver various payloads (e.g. anti-cancer drugs, proteins, DNA or siRNA) to targeted cells.4–11 Following systemic administration, nanoparticles can accumulate in the liver in less than a few minutes following intravenous injection, primarily due to the opsonization process and discontinuous endothelium of the microvessels in the liver.12 However, hepatic filtration is an important biological barrier to nanoparticles, in which phagocytic Kupffer cells play the most important role in the hepatic clearance of nanoparticles. Thus, most nanoparticles may be primarily cleared by Kupffer cells and not internalized by hepatocytes upon accumulation in liver tissue. To solve this problem, many studies have utilized ligands to modify nanoparticles, aiming to improve the targeting ability to hepatocytes or to hepatocellular carcinoma (HCC) cells.13,14 Galactosyl ligands are well-known to target the asialoglycoprotein receptor (ASGP-R), a lectin predominantly expressed on the sinusoidal surface of mammalian hepatocytes.15 It has also been well demonstrated that the interaction between galactosyl ligands with ASGP-R mediates enhanced the endocytosis of nanoparticles by hepatocytes,16–20 and drug delivery systems with the modification of galactosyl ligands are useful for the drug delivery to liver in vivo.16,21–28
Herein, we developed a core-cross-linked polyphosphoester nanogel surrounded by galactosylated poly(ethylene glycol) (PEG) arms, and loaded doxorubicin (DOX) into the nanogel to target hepatocytes and hepatocellular carcinoma cells. We studied the overall localization of the DOX-loaded nanogel in the liver and compared the internalization of galactosylated and non-galactosylated nanoparticles by hepatocytes and Kupffer cells following intravenous injection. The impact of the DOX-loaded nanogel bearing galactosyl ligands on the therapeutic efficacy in diethylnitrosamine (DEN)-induced HCC was further investigated in rats. The results demonstrated that galactosyl ligands on nanogel increased hepatic internalization and improved therapeutic efficiency of HCC in vivo.
2 Materials and methods
2.1 Materials
3,6-Dioxaoctan-1,8-diyl bis(ethylene phosphate) (TEGDP) was synthesized as previously described.29 α-Hydroxy-ω-azido poly(ethylene glycol) (HO-PEG3400-N3, Mw = 3400) was synthesized according to the literature.30 Poly(ethylene glycol) monomethyl ether (mPEG2000, Mw = 2000, Acros Organics) and HO-PEG3400-N3 were dried twice by azeo distillation of toluene before uses. Stannous octoate (Sn(Oct)2, Sinopharm Chemical Reagent Co., China) was purified according to a method described in the literature.31 Dimethyl sulfoxide (DMSO) was refluxed with calcium hydride and distilled before use. 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) and N-hydroxysulfosuccinimide sodium salt (Sulfo-NHS) were bought from Shanghai Medpep Co. (Shanghai, China). All other chemicals for syntheses were of reagent grade and used without further purification.Doxorubicin hydrochloride was purchased from Zhejiang Hisun Pharmaceutical Co. Diethylnitrosamine and lactobionic acid were purchased from Sigma-Aldrich Co. DAPI and Alexa Fluor® 488 phalloidin were purchased from Invitrogen Life Science.
2.2 Animals
Male Wistar rats (125–150 g) were purchased from Experimental Animal Center, Chinese Science Academy (Shanghai, China). All animals received care in compliance with the guidelines outlined in the Guide for the Care and Use of Laboratory Animals and the procedures were approved by the University of Science and Technology of China Animal Care and Use Committee.2.3 Syntheses of non-galactosylated nanogel (m-PPE) and galactosylated nanogel (Gal-PPE)
The nanogels were synthesized according to the procedure reported by us previously.29 To obtain mPEG-armed and polyphosphoester core-cross-linked m-PPE (Scheme S1†), mPEG2000 (1 g, 0.5 mmol), TEGDP (0.79 g, 3 mmol) and 6 mL of anhydrous DMSO were added into a fresh flamed and nitrogen purged round-bottomed flask in a glove box with H2O and O2 contents less than 0.1 ppm. After stirring at 60 °C for 30 min to allow complete dissolution of mPEG2000, Sn(Oct)2 (20 mg, 0.05 mmol) was added quickly. The reaction was carried out at 60 °C for an additional 12 h. The product was diluted with Milli-Q water and dialyzed against water for 3 days using a Spectra/Por® dialysis membrane (MWCO 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000). m-PPE was obtained after lyophilization with a yield of approximately 65%.
000). m-PPE was obtained after lyophilization with a yield of approximately 65%.
To synthesize Gal-PPE (Scheme S2†), a mixture of mPEG2000 and HO-PEG3400-N3 at a molar ratio of 1:1 was used as the macroinitiator, and the reaction and purification were performed similarly as described above. The resulting nanogel (500 mg) was then dissolved in 5 mL of tetrahydrofuran and the azido groups of nanogel were reduced to amino groups with triphenylphosphine (10 mg, 0.038 mmol) at 25 °C for 4 h, followed by the addition of water (100 μL) and stirring overnight. The product, which was an amino-functionalized nanogel, was obtained by precipitation in cold diethyl ether (100 mL) twice. In the next step, the resulting amino-functionalized nanogel (100 mg) was dissolved in 50 mL of 2-(4-morpholino) ethanesulfonic acid buffer (MES buffer, 0.1 M, pH 6.5), to which was added Sulfo-NHS and EDC activated lactobionic acid (5 mmol, Sigma) in 50 mL of MES buffer. The reaction was performed at 4 °C for 12 h and at room temperature for another 12 h. The product was purified by dialysis against Milli-Q water for 3 days using a Spectra/Por® dialysis membrane tubing with MWCO 2000, and lyophilized overnight.
2.4 Physicochemical characterizations
A Bruker AV300 NMR spectrometer (300 MHz) was used for NMR measurements. Deuterated chloroform containing 0.03% tetramethylsilane was used as the solvent. Elementar Vario ELIII was used to measure the content of C and H elements of the nanogel. An Analyst 800 spectrometer was used to measure the content of P element of the nanogel.Transmission electron microscopy measurements (TEM) were performed on a JEOL-2010 transmission electron microscope with an accelerating voltage of 200 kV. The samples were prepared by pipetting onto 230 mesh copper grids coated with carbon and allowing the sample to dry in air before measurements.
The size and size distribution of the nanogel in aqueous solution were measured by dynamic light scattering, carried out on a Malvern Zetasizer Nano ZS90 with a He–Ne laser (633 nm) and 90° collecting optics. The data were analyzed by Malvern Dispersion Technology Software 4.20.
2.5 Drug loading
m-PPE or Gal-PPE (100 mg) was mixed with doxorubicin hydrochloride in 1 mL of Milli-Q ultrapurified water at a weight ratio of 10:1 (nanogel to DOX). The mixture was stirred gently for 3 days in the dark. Samples were taken out and ultra-filtrated by an Amicon® Ultra-4 centrifugal filter. The concentration of DOX in the filtration was determined using high performance liquid chromatography (HPLC) as described previously.32 The DOX-loaded nanogels (further denoted as m-PPE–DOX and Gal-PPE–DOX) were finally obtained by purification with sepharose CL-4B gel and lyophilization.
2.6 Galactosyl ligand-mediated internalization in vitro
HepG2 cells from the American Type Culture Collection (ATCC) were cultured with RPMI 1640 (Invitrogen) supplemented with penicillin (100 units per mL), streptomycin (100 μg mL−1) and 10% heat-inactivated fetal bovine serum (FBS, Hyclone) in a humidified atmosphere of 5% CO2 at 37 °C. Cells were plated into a 24-well plate at a density of 1.5 × 105 cells per well, and the culture medium was replaced with fresh complete culture medium containing either m-PPE–DOX or Gal-PPE–DOX, which were precooled to 4 °C. The final concentration of DOX cultured with cells was equivalent to 20 μg mL−1. In the competitive inhibition assay, lactobionic acid at a final concentration of 30 or 60 mM was added to the complete culture medium. The cells were cultured at 4 °C for 2 h, and washed twice by precooled PBS (0.01 M, pH 7.4) at 4 °C. Fresh complete culture medium at 37 °C was then added and the cells were cultured for an additional 2 h at 37 °C. The cells were finally trypsinized, washed with PBS twice, resuspended in 200 μL of PBS and subjected to flow cytometric analyses using a Becton Dickinson FACSCalibur flow cytometer. Galactosyl ligand-mediated internalization by rat hepatocytes or co-cultured rat hepatocytes and rat Kupffer cells was determined similarly as described above. The isolation of rat hepatocytes or rat Kupffer cells is described in the ESI.† Rat hepatocytes (6 × 105 per well) or mixed rat hepatocytes and rat Kupffer cells (6 × 105 and 1 × 105 per well, respectively) were plated into a 6-well plate. Cells were cultured for 24 h before treatments.2.7 Analyses of the internalization of DOX or DOX-loaded nanoparticles by hepatocytes following i.v. injection
Rats received a single intravenous injection of free DOX, m-PPE–DOX or Gal-PPE–DOX at an equivalent DOX dose of 10 μg g−1 of body weight. The rats were then sacrificed at a designated time. Hepatocytes were separated from rat livers as previously described with a modification.33Liver tissues were perfused with an SC-1 solution (consisting of 8000 mg L−1 NaCl, 400 mg L−1 KCl, 88.17 mg L−1 NaH2PO4·2H2O, 120.45 mg L−1 Na2HPO4, 2380 mg L−1 HEPES, 350 mg L−1 NaHCO3, 190 mg L−1 EGTA, 900 mg L−1 glucose, pH 7.3) for 5 min until the liver turned khaki. The liver tissues were further perfused with 0.05% collagenase dissolved in an SC-2 solution (consisting of 8000 mg L−1 NaCl, 400 mg L−1 KCl, 88.17 mg L−1 NaH2PO4·2H2O, 120.45 mg L−1 Na2HPO4, 2380 mg L−1 HEPES, 350 mg L−1 NaHCO3, 560 mg L−1 CaCl2·2H2O, pH 7.3) for 12 min at 37 °C. After perfusion, the liver was dispersed with forceps in an ice-cold SC-2 solution and filtered through a 150-gauge nylon gauze. The cell suspension was centrifuged at 50 g for 10 min at 4 °C, and the cell pellet was washed and centrifuged at 50 g for 5 min three times in PBS (0.01 M, pH 7.4). To separate the hepatocytes from the total cells, the pellet was resuspended and centrifuged at 400 g for 10 min using 40% Percoll. The cells at the bottom were the hepatocytes and were washed and centrifuged at 250 g for 3 min. The hepatocytes were resuspended by cold PBS, and analyzed using a Becton Dickinson FACSCalibur flow cytometer.
2.8 Biodistribution of DOX-loaded nanogels and hepatic location
To observe DOX distribution, animals were administered with free DOX, m-PPE–DOX or Gal-PPE–DOX at an equivalent DOX dose of 10 μg g−1 of body weight. After 24 h, rats were sacrificed and the liver tissues were sectioned (7 μm thick) by a cryostat (Leica CM1900, Leica Microsystems). Sections were counterstained with DAPI for the cell nucleus and Alexa Fluor® 488 phalloidin for the cell membrane following the manufacturer's instructions. All the sections were imaged using a Zeiss LSM710 laser confocal scanning microscopy imaging system.2.9 Inducement of hepatocellular carcinoma (HCC) in rats and treatments
HCC was induced in male Wistar rats with DEN, given in their drinking water at 100 mg L−1 for 10 weeks. Two weeks after the last day of DEN administration, animals were randomly assigned, and administrated with PBS, free DOX, m-PPE–DOX or Gal-PPE–DOX respectively at DOX level of 1 μg g−1 of body weight by tail intravenous injection every four days, in a volume of 1 μL g−1 weight. Eight administrations were performed. The survival of the rats was observed during the 160 days after the first administration.In a separate experiment, one week after the last administration, rats were sacrificed under isoflurane anesthesia. The liver tissues were removed and the lobes were separated. Tumor nodules larger than 3 mm in diameter were counted on the surface of each lobe, and the difference in the nodule numbers in four experimental groups was statistically evaluated.
2.10 Statistical analysis
Student's t test was performed in analyses of doxorubicin localization and hepatic distribution measurements. The Kruskal–Wallis test was used to compare the number of neoplastic nodules between groups. A Kaplan–Meier analysis and log-rank test were performed between groups in survival analyses.3 Results and discussion
3.1 Preparation and characterization of doxorubicin-loaded nanogels
We employed a one-step ring-opening polymerization process to synthesize the nanogel as the delivery system.29 The obtained nanogels contained core-cross-linked polyphosphoester and were surrounded by either monomethyl poly(ethylene glycol) (m-PEG) or galactosylated poly(ethylene glycol) (Gal-PEG) arms. The nanogels were further denoted as m-PPE and Gal-PPE, respectively. The structures of the nanogels are illustrated in Fig. 1A.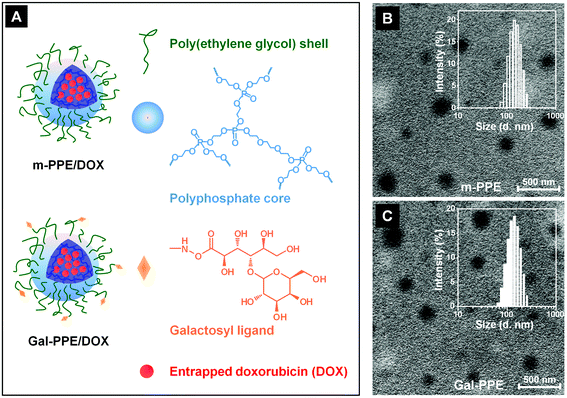 | ||
| Fig. 1 (A) Schematic illustration of m-PPE and galactosylated Gal-PPE loaded with doxorubicin (DOX); (B and C) transmission electron microscopy images of m-PPE (B) and Gal-PPE (C). The insets in B and C represent the size and size distributions in water measured by dynamic light scattering. | ||
Both m-PPE and Gal-PPE could be well-dispersed in water, exhibiting clear core–shell structures with average diameters around 130 nm, as demonstrated by transmission electron microscopy observations, which were consistent with those measured by dynamic light scattering (Fig. 1B and 1C). Gal-PPE contained 32.4 mol% galactosylated poly(ethylene glycol) arms according to elemental analyses. 1H NMR analyses shown in the ESI (Fig. S1†) demonstrated the successful synthesis of Gal-PPE nanogels, exhibiting resonances at 2.7–3.3 ppm, which were assigned to protons of the lactosyl groups. It was noteworthy that the surface zeta potentials of m-PPE and Gal-PPE in water were very similar, being −19.3 and −18.4 mV, respectively. Our previous study demonstrated that such nanogels could be used to encapsulate DOX efficiently, simply by incubating the drug with the nanogels.29 In this study, the amount of DOX loaded into the Gal-PPE was 8.3 wt% with around 85% loading efficiency, while the efficiency of drug loading into the m-PPE was only slightly lower at 7.1 wt%. The DOX-loaded nanogels were further denoted as m-PPE–DOX and Gal-PPE–DOX. Release profiles of DOX from m-PPE–DOX and Gal-PPE–DOX in phosphate buffered saline at pH 7.4 and 37 °C indicated that around 17% of the overall loaded DOX was released in around 30 h and there was no significant difference between m-PPE–DOX and Gal-PPE–DOX (ESI, Fig. S2†). Moreover, this nanogel with a reduced DOX release in normal phosphate buffered saline might be advantageous in systemic administration, since the drug release can be accelerated in cells considering the presence of phosphodiesterase I enzyme in mammalian cells.
3.2 Cellular uptake of DOX-loaded Gal-PPE nanogels
To demonstrate the specific binding of Gal-PPE–DOX to mammalian hepatocytes via the interaction of galactosyl ligands with ASGP-R, we incubated HepG2 cells with either Gal-PPE–DOX or m-PPE–DOX at 4 °C for 2 h to allow binding but minimized internalization, since it is well-known that HepG2 cells express ASGP-R on the membrane. After washing off the unbound nanoparticles, the cells were further cultured at 37 °C for 1 h and analyzed by flow cytometry. The results demonstrated that the fluorescence intensity was much higher in cells treated with Gal-PPE–DOX than in those treated with m-PPE–DOX, implying the presence of a specific interaction of Gal-PPE–DOX with the cell surface (Fig. 2A). The interaction of the galactosyl ligands of Gal-PPE–DOX with the ASGP-R on the cells was further confirmed by a competitive inhibition assay. When hepatocytes were incubated with m-PPE–DOX, no significant difference was observed in the cellular fluorescence intensity regardless of the addition of a lactobionic acid inhibitor to the medium (Fig. 2B). However, when the cells were incubated with Gal-PPE–DOX, cellular fluorescence intensity was dependent on the concentration of lactobionic acid (Fig. 2C). Higher concentrations of lactobionic acid decreased the cellular fluorescence intensity more efficiently, suggesting that lactobionic acid competitively prevented the interaction of Gal-PPE–DOX with the cells, thus demonstrating that cell-surface ASGP-R mediated the internalization of Gal-PPE–DOX.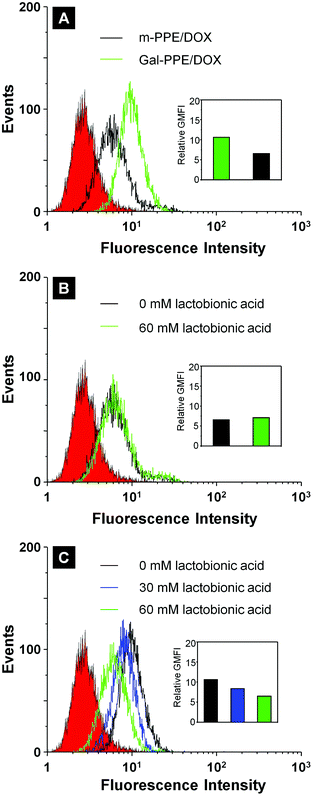 | ||
| Fig. 2 Cellular fluorescence intensity of HepG2 cells incubated with m-PPE–DOX or Gal-PPE–DOX in the absence (A) or presence of a lactobionic acid inhibitor (B and C, with m-PPE–DOX or Gal-PPE–DOX, respectively). The relative geometrical mean fluorescence intensities (GMFI) of cells are shown as insets. | ||
The specific interaction between galactosyl ligands of Gal-PPE with ASGP-R was further demonstrated in the incubation of Gal-PPE with rat primary hepatocytes. As shown in Fig. 3A, the fluorescence intensity was much higher in rat hepatocytes treated with Gal-PPE–DOX than in those treated with m-PPE–DOX by an identical approach described above. Moreover, the interaction of galactosyl ligands of Gal-PPE–DOX with the ASGP-R on the cells was also hindered by the addition of lactobionic acid. However, such an inhibition effect was not significant when the rat hepatocytes were cultured with m-PPE–DOX.
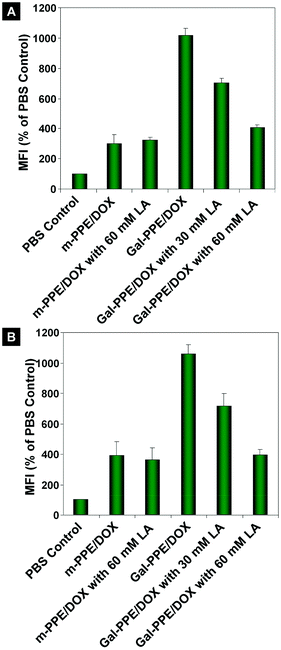 | ||
| Fig. 3 The relative geometrical mean fluorescence intensities (GMFI) of DOX in hepatocytes after incubation with m-PPE–DOX or Gal-PPE–DOX at the equivalent dose of DOX. Lactobionic acid (LA) was used as the inhibitor in the cell culture. m-PPE–DOX or Gal-PPE–DOX was cultured with either rat hepatocytes (A), or with co-cultured rat hepatocytes and Kupffer cells at a cell number ratio of 6:1 (B). | ||
In another experiment, the impact of galactosyl ligands of Gal-PPE–DOX on the internalization by rat hepatocytes was further validated by incubating Gal-PPE–DOX with co-cultured rat hepatocytes and Kupffer cells at a cell number ratio of 6:1. The results shown in Fig. 3B revealed that the cellular internalization patterns of m-PPE–DOX and Gal-PPE–DOX by rat hepatocytes in the co-culture system were not significantly changed. Although it was hard to directly compare the amounts of Gal-PPE–DOX internalized by rat hepatocytes and Kupffer cells in the co-culture system, these results demonstrated that the interaction between galactosyl ligands of Gal-PPE with the ASGP-R on the surface of rat hepatocytes was not hindered by the presence of Kupffer cells.
3.3 Biodistribution and hepatic location of Gal-PPE–DOX
We next determined the distribution of DOX in hepatocytes after the administration of the different formulations. The rats received a single intravenous injection of free DOX, m-PPE–DOX or Gal-PPE–DOX at an equivalent dose of DOX. Liver tissues were disaggregated and suspended, and the population of hepatocytes was distinguished via cytometric detection of the DOX fluorescence in both cells. As shown in Fig. 4, in comparison with the free DOX injection, the fluorescence intensities of DOX in rat hepatocytes were significantly stronger at 12 or 24 h when the rats received the m-PPE–DOX or Gal-PPE–DOX injections. Moreover, with the delivery of Gal-PPE–DOX, the fluorescence intensity of DOX in rat hepatocytes was more pronounced at 24 h when it was compared with free DOX or the m-PPE–DOX injection. This indicated that the modification of the nanogel with hepatocyte-specific galactosyl ligands had a significant impact on DOX accumulation in hepatocytes.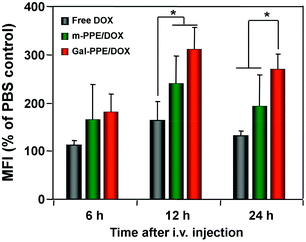 | ||
| Fig. 4 The relative geometrical mean fluorescence intensities (GMFI) of DOX in rat hepatocytes. Rats received a single intravenous injection of free DOX, m-PPE–DOX or Gal-PPE–DOX at an equivalent DOX dose of 10 μg g−1 of body weight (n = 3 per group). Student's t test was performed. * p < 0.05. | ||
In order to further demonstrate the distribution of DOX-loaded nanoparticles at the cellular level, liver tissues were collected and sectioned 24 h after intravenous administration. The cell nucleus and cell membranes were counterstained with DAPI and Alexa Fluor® 488 phalloidin, respectively. As shown in Fig. 5, the parenchymal cells demonstrated polyhedral morphology with regular, round nuclei and represented about 60% of the cells in the liver, but almost 80% of the hepatic volume. Kupffer cells, or hepatic macrophages, demonstrated irregular nuclei and were generally located in the lumen of the hepatic sinusoids adjacent to hepatocytes, and were in direct contact with the bloodstream and represented about 35% of the cells in the liver.34 From Fig. 5, DOX was preferentially located in non-parenchymal cells rather than in parenchymal cells when free DOX or m-PPE–DOX was administrated, in contrast to the administration of Gal-PPE–DOX, which was associated with significant cellular uptake by parenchymal cells.
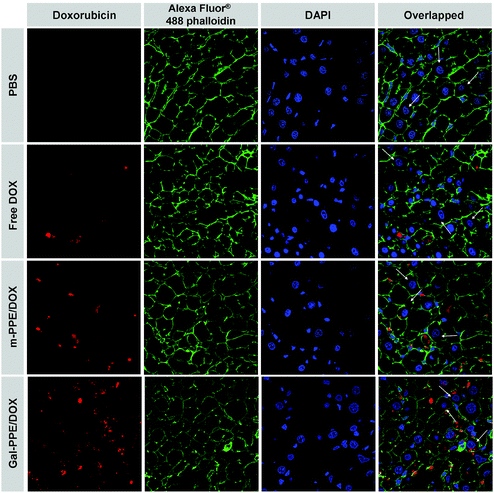 | ||
| Fig. 5 Cellular localization patterns of DOX in liver tissue following a single administration of free DOX, m-PPE–DOX or Gal-PPE–DOX to rats at an equivalent DOX dose of 10 μg g−1 of body weight. The cell nuclei were counterstained with DAPI (blue), the cell membranes were stained with Alexa Fluor® 488 phalloidin (green) and the doxorubicin was shown in red. White arrows indicate hepatocytes. | ||
3.4 Anti-HCC activity of Gal-PPE–DOX in vivo
With similar overall localization in the liver, increased hepatocyte uptake of Gal-PPE–DOX should represent a robust advantage in the treatment of liver disease. To demonstrate it, we established primary HCC in rat liver using DEN as a carcinogen.35 It must be mentioned that DEN favors HCC development from hepatocytes, and that this chemically-induced HCC animal model is very similar to the injury-fibrosis-malignancy cycle which occurs in humans.14,36 We treated HCC-bearing animals with PBS, free DOX, m-PPE–DOX or Gal-PPE–DOX, respectively, every four days, at a DOX level of 1 μg g−1 of body weight by intravenous tail injection on eight occasions. In one experiment, one week after the last administration, rats were sacrificed under isoflurane anesthesia. Liver tissues were removed and the lobes were separated. Tumor nodules larger than 3 mm in diameter were counted on the surface of each lobe, and the difference in nodule number in the four experimental groups was statistically evaluated. In another experiment, the survival rate of HCC-bearing rats after eight administrations was recorded.Fig. 6A shows that the number of neoplastic nodules was significantly decreased in rats when the animals were treated with Gal-PPE–DOX. At the same time, significant numbers of malignant nodules were observed on the surface of the livers from HCC-bearing rats receiving PBS, free DOX or m-PPE–DOX, while the livers of HCC-bearing rats treated with Gal-PPE–DOX exhibited inconspicuous and smaller nodules (ESI, Fig. S3†). More importantly, the survival rate of HCC-bearing rats after eight administrations demonstrated a significantly improved therapeutic efficacy when DOX was delivered by the Gal-PPE nanogel (Fig. 6B). Eight of the ten rats in the Gal-PPE–DOX treated group remained alive after 160 days from the first administration, while all the rats given PBS or free DOX treatments died. A significant survival difference was also observed between the groups receiving the Gal-PPE–DOX and m-PPE–DOX treatments (log-rank = 0.0179).
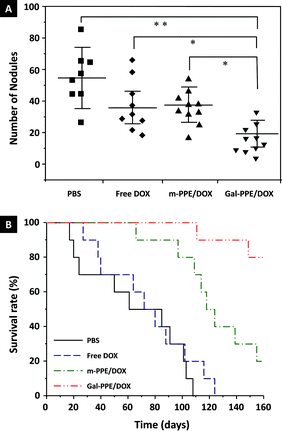 | ||
| Fig. 6 Anti-HCC efficacy of DOX-loaded nanogels. (A) Number of neoplastic nodules (≥3 mm in diameter) counted on the liver of each rat. Two and one rats died in the PBS and free DOX treatment groups during the treating period, respectively. The statistical significance was evaluated by the non-parametric Kruskal–Wallis test (*p < 0.005; **p < 0.001). (B) Survival analysis of HCC-bearing rats after treatment with PBS, free DOX, m-PPE–DOX or Gal-PPE–DOX. | ||
Although it has been clearly demonstrated that the inclusion of targeting molecules in nanoparticle delivery systems can result in increased tumor treatment efficacy, and the impact of tumor-targeting moieties associated with nanoparticles has recently been addressed,37–39 nanoparticles following systemic administration have not been well understood. It is not doubted that nanoparticles are a complicated and multifactorial issue. For example, the size and size distribution, surface zeta potential, chemical composition and PEGylation of nanoparticles may play important roles on their fate in vivo.4,40,41 In this study, to minimize the differences between liganded and unliganded nanoparticles, we synthesized core–shell m-PPE and Gal-PPE nanogels, which possessed similar compositions, equal size and size distributions and similar surface zeta potentials. More importantly, both m-PPE and Gal-PPE nanogels were PEGylated, which would potentially prevent serum protein binding and rapid clearance by the reticuloendothelial system. This study demonstrates that modification of hydrogel nanoparticles with hepatocyte-specific ligands impacts their recognition, cellular uptake by hepatocytes and the efficacy of drug-loaded nanoparticles.
4 Conclusions
In summary, we synthesized core–shell polyphosphoester nanogels, m-PPE and Gal-PPE, which were surrounded by poly(ethylene glycol) (PEG) arms without or with tethered hepatocyte-specific galactosyl ligands, respectively. The m-PPE and Gal-PPE nanogels possessed similar compositions, equal size and size distributions, similar surface zeta potentials, and exhibited comparable DOX loading capacities. It was demonstrated that the galactosyl ligands of Gal-PPE nanogels mediated specific cell internalization by interaction with ASGP-R in cell culture, leading to increased accumulation of DOX-loaded Gal-PPE nanoparticles in ASGT-R expressing HepG2 cells, in comparison with DOX-loaded m-PPE nanoparticles. Further studies revealed that the liganded Gal-PPE nanoparticles were taken up more by hepatocytes in rats’ liver following systemic injection, compared with unliganded m-PPE nanoparticles. Meantime, a galactosylated modification of the nanogels promoted the therapeutic efficacy in treatments of diethylnitrosamine-induced HCC in rats. The results revealed that a targeting modification of nanoparticle drug carriers has the potential to overcome intrinsic biological barriers, even hepatic clearance, and therefore improve the therapeutic efficacy of drugs.Acknowledgements
This work was supported by the National Basic Research Program of China (973 Programs, 2012CB932500 and 2010CB934001), the National Natural Science Foundation of China (51125012, 51203145), the Fundamental Research Funds for the Central Universities (WK2070000008) and the Open Project of State Key Laboratory of Supramolecular Structure and Materials (SKLSSM201301).Notes and references
- Z. H. Xu, L. L. Chen, W. W. Gu, Y. Gao, L. P. Lin, Z. W. Zhang, Y. Xi and Y. P. Li, Biomaterials, 2009, 30, 226–232 CrossRef CAS.
- J. Shi, A. R. Votruba, O. C. Farokhzad and R. Langer, Nano Lett., 2010, 10, 3223–3230 CrossRef CAS.
- S. Acharya and S. K. Sahoo, Adv. Drug Delivery Rev., 2011, 63, 170–183 CrossRef CAS.
- O. Veiseh, J. W. Gunn and M. Zhang, Adv. Drug Delivery Rev., 2010, 62, 284–304 CrossRef CAS.
- D. J. Naczynski, T. Andelman, D. Pal, S. Chen, R. E. Riman, C. M. Roth and P. V. Moghe, Small, 2010, 6, 1631–1640 CrossRef CAS.
- F. M. Kievit, O. Veiseh, C. Fang, N. Bhattarai, D. Lee, R. G. Ellenbogen and M. Zhang, ACS Nano, 2010, 4, 4587–4594 CrossRef CAS.
- A. Potineni, D. M. Lynn, R. Langer and M. M. Amiji, J. Controlled Release, 2003, 86, 223–234 CrossRef CAS.
- J. Z. Du, T. M. Sun, W. J. Song, J. Wu and J. Wang, Angew. Chem., Int. Ed., 2010, 49, 3621–3626 CrossRef CAS.
- M. De, P. S. Ghosh and V. M. Rotello, Adv. Mater., 2008, 20, 4225–4241 CrossRef CAS.
- D. Kim, E. S. Lee, K. T. Oh, Z. G. Gao and Y. H. Bae, Small, 2008, 4, 2043–2050 CrossRef CAS.
- D. P. Ferris, J. Lu, C. Gothard, R. Yanes, C. R. Thomas, J. C. Olsen, J. F. Stoddart, F. Tamanoi and J. I. Zink, Small, 2011, 7, 1816–1826 CrossRef CAS.
- L. Grislain, P. Couvreur, V. Lenaerts, M. Roland, D. Deprez-Decampeneere and P. Speiser, Int. J. Pharm., 1983, 15, 335–345 CrossRef CAS.
- H. F. Liang, C. T. Chen, S. C. Chen, A. R. Kulkarni, Y. L. Chiu, M. C. Chen and H. W. Sung, Biomaterials, 2006, 27, 2051–2059 CrossRef CAS.
- L. Fiume, L. Bolondi, C. Busi, P. Chieco, F. Kratz, M. Lanza, A. Mattioli and G. Di Stefano, J. Hepatol., 2005, 43, 645–652 CrossRef CAS.
- G. Ashwell and J. Harford, Annu. Rev. Biochem., 1982, 51, 531–554 CrossRef CAS.
- B. G. Davis and M. A. Robinson, Curr. Opin. Drug Discovery Dev., 2002, 5, 279–288 CAS.
- J. C. Perales, G. A. Grossmann, M. Molas, G. Liu, T. Ferkol, J. Harpst, H. Oda and R. W. Hanson, J. Biol. Chem., 1997, 272, 7398–7407 CrossRef CAS.
- P. Midoux, C. Mendes, A. Legrand, J. Raimond, R. Mayer, M. Monsigny and A. C. Roche, Nucleic Acids Res., 1993, 21, 871–878 CrossRef CAS.
- K. Sagara and S. W. Him, J. Controlled Release, 2002, 79, 271–281 CrossRef CAS.
- T. Eto and H. Takahashi, Nat. Med., 1999, 5, 577–581 CrossRef CAS.
- S. Kawakami, S. Fumoto, M. Nishikawa, F. Yamashita and M. Hashida, Pharm. Res., 2000, 17, 306–313 CrossRef CAS.
- C. S. Cho, K. Y. Cho, I. K. Park, S. H. Kim, T. Sasagawa, M. Uchiyama and T. Akaike, J. Controlled Release, 2001, 77, 7–15 CrossRef CAS.
- P. C. N. Rensen, L. A. Sliedregt, M. Ferns, E. Kieviet, S. M. W. van Rossenberg, S. H. van Leeuwen, T. J. C. van Berkel and E. A. L. Biessen, J. Biol. Chem., 2001, 276, 37577–37584 CrossRef CAS.
- Y. Hattori, S. Kawakami, F. Yamashita and M. Hashida, J. Controlled Release, 2000, 69, 369–377 CrossRef CAS.
- C. S. Cho, A. Kobayashi, R. Takei, T. Ishihara, A. Maruyama and T. Akaike, Biomaterials, 2000, 22, 45–51 CrossRef.
- S. Kawakami, C. Munakata, S. Fumoto, F. Yamashita and M. Hashida, J. Pharm. Sci., 2001, 90, 105–113 CrossRef CAS.
- I. K. Park, Y. H. Park, B. A. Shin, E. S. Choi, Y. R. Kim, T. Akaike and C. S. Cho, J. Controlled Release, 2000, 69, 97–108 CrossRef CAS.
- L. A. Sliedregt, P. C. Rensen, E. T. Rump, P. J. van Santbrink, M. K. Bijsterbosch, A. R. Valentijn, G. A. van der Marel, J. H. van Boom, T. J. van Berkel and E. A. Biessen, J. Med. Chem., 1999, 42, 609–618 CrossRef CAS.
- M. H. Xiong, J. Wu, Y. C. Wang, L. S. Li, X. B. Liu, G. Z. Zhang, L. F. Yan and J. Wang, Macromolecules, 2009, 42, 893–896 CrossRef CAS.
- J. Li and W. J. Kao, Biomacromolecules, 2003, 4, 1055–1067 CrossRef CAS.
- K. Stridsberg, M. Ryner and A. C. Albertsson, Macromolecules, 2000, 33, 2862–2869 CrossRef CAS.
- J. Wu, X. Q. Liu, Y. C. Wang and J. Wang, J. Mater. Chem., 2009, 19, 7856–7863 RSC.
- D. B. Kristensen, N. Kawada, K. Imamura, Y. Miyamoto, C. S. TatenoSeki, T. Kuroki and K. Yoshizato, Hepatology, 2000, 32, 268–277 CrossRef CAS.
- Y. Adachi, B. U. Bradford, W. Gao, H. K. Bojes and R. G. Thurman, Hepatology, 1994, 20, 453–460 CrossRef CAS.
- M. F. Rajewsky, W. Dauber and H. Frankenberg, Science, 1996, 152, 83–85 Search PubMed.
- H. Nagata, E. Hatano, M. Tada, M. Murata, K. Kitamura, H. Asechi, M. Narita, A. Yanagida, N. Tamaki, S. Yagi, I. Ikai, K. Matsuzaki and S. Uemoto, Hepatology, 2009, 49, 1944–1953 CrossRef CAS.
- C. Mamot, D. C. Drummond, C. O. Noble, V. Kallab, Z. X. Guo, K. L. Hong, D. B. Kirpotin and J. W. Park, Cancer Res., 2005, 65, 11631–11638 CrossRef CAS.
- D. B. Kirpotin, D. C. Drummond, Y. Shao, M. R. Shalaby, K. Hong, U. B. Nielsen, J. D. Marks, C. C. Benz and J. W. Park, Cancer Res., 2006, 66, 6732–6740 CrossRef CAS.
- D. W. Bartlett, H. Su, I. J. Hildebrandt, W. A. Weber and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 15549–15554 CrossRef CAS.
- S. Uchida, K. Itaka, Q. X. Chen, K. Osada, K. Miyata, T. Ishii, M. Harada-Shiba and K. Kataoka, J. Controlled Release, 2011, 155, 296–302 CrossRef CAS.
- Y. Hattori, S. Kawakami, F. Yamashita and M. Hashida, J. Controlled Release, 2000, 69, 369–377 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3bm60099h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2013 |