Biomaterials in RNAi therapeutics: quo vadis?
Ernst
Wagner
*
Pharmaceutical Biotechnology, Center for System-Based Drug Research, Department of Pharmacy and Center for Nanoscience (CeNS), Ludwig-Maximilians-University, Butenandtstrasse 5-13, 81377 Munich, Germany. E-mail: ernst.wagner@cup.uni-muenchen.de; Fax: +49 89 2180 77791; Tel: +49 89 2180 777841
First published on 10th May 2013
Abstract
The fifteen years of utilizing RNAi present a surprise story, ranging from the unexpected discovery and publication of RNA interference in 1998, rewarded by the nobel prize in 2006, the introduction of synthetic short siRNAs for the specific gene silencing in mammalian cells in 2001, or the discovery of more than 1600 human microRNAs naturally regulating about one third of our genes. Therapeutic applications started amazingly fast and resulted in the first recent successes in therapy. Synthetic siRNAs are under evaluation for knocking down disease-associated target mRNAs, microRNA mimics for turning on or antagonists (antagomirs) for turning off microRNA activity. Modified oligonucleotides comprise a special class of therapeutics with a new chemical profile; the precise synthetic molecules are much smaller than protein or gene vector drugs, but they are larger than conventional drugs and thus cannot passively diffuse into their target cells. The main current strategies for solving the delivery problem are discussed. We now face the interesting question of alternative future directions: should oligonucleotide molecules be chemically further minimized into small drug-like chemical entities? Or should multiple RNAi molecules be wrapped up into larger virus-like nanoparticles for delivery? Biomaterials in therapeutic RNA interference, quo vadis?
1. Introduction
The discovery of RNA interference (RNAi)1 has fundamentally changed our knowledge in biology about gene regulation. Small double-stranded RNA molecules, either derived from our endogenous >1600 human microRNA genes2 or artificially introduced from outside, are processed into a so-called RISC complex, which have one (‘guide strand’) of the two 21–23 nucleotide long RNA strands incorporated in phosphorylated form complexed with argonaute (Ago) protein.3 These RISC complexes similarly as antisense oligonucleotides recognize in the intracellular cytosol complementary target messenger RNA (mRNA) and prevent protein translation. Natural human microRNA RISCs4,5 contain predominantly argonaute Ago1 or Ago2 and regulate about one third of our genes. The sequence match with the target mRNA is only partial and is strictly required for the first 7–8 nucleotides (seed region); these microRNA RISCs interfere by hybridization and sterically blocking mRNA against translation; subsequent mRNA degradation occurs by other processes. In contrast, siRNA RISCs containing Ago2 with an endonuclease domain can cleave perfectly matched complementary mRNA in a catalytic fashion. In mammals this process is not naturally occurring, but can be artificially triggered by the introduction of synthetic short siRNA molecules.6This enhanced understanding of gene regulation has opened our mind for new medical concepts (Fig. 1).7–9 Novel therapeutics based on synthetic siRNAs can be utilized to silence malfunctioning or disease-promoting genes. Introduction of microRNA mimics of ‘tumor suppressor’ miRs for turning on therapeutic microRNA activity displays encouraging anticancer activity.10,11 Conversely, single-stranded oligonucleotides complementary to microRNA RISCs may act as antagonists (antagomirs)12 for turning off tumor-associated ‘onco’ microRNA activity. Altogether, RNAi therapeutic modulators comprise a new class of medium-sized therapeutics based on synthetic single- or double-stranded modified RNA oligonucleotides. For broader medical translation, delivery to their intracellular cytosolic site of drug action is the key current limitation, as these molecules cannot passively diffuse into their target cells.13 In the following, diverse strategies for overcoming the roadblocks of delivery are reviewed.
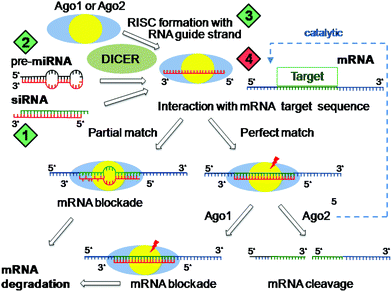 | ||
| Fig. 1 RNA interference modes of action. Incorporation of guide RNA strand (derived from natural microRNA or introduced siRNA) together with argonaute (Ago1 or Ago2) into the RNA interference silencing complex (RISC). RISCs recognize complementary messenger RNA (mRNA) strands by hybridization, which always matches in the seed region (the first 7–8 nucleotides of 5′-end of the guide strand). In the case of a perfect sequence match over the whole guide strand, Ago2 RISCs (which contain an endonucleolytic RNase activity) cleave the target mRNA in a catalytic mode, whereas RISCs with the partial sequence match repress protein translation by mRNA blockade. Green diamonds present options for artificial induction of RNA interference (1: synthetic siRNA; 2: synthetic pre-microRNA; 3: single stranded ss-siRNA), the red diamond presents options for inhibition of RNA interference (4: antagomirs such as tiny-LNA, masking the RISC guide strand by hybridization). | ||
2. Strategies to maximize cytosolic delivery
The main roadblocks against efficient cytosolic siRNA delivery are as follows: (i) Double-stranded siRNA oligonucleotides are too charged, too large and too rigid to migrate across cellular membranes. (ii) They are however small enough to be rapidly removed from blood circulation by renal clearance. (iii) They are biologically vulnerable and degraded in the extracellular and intracellular biological environments. (iv) As negatively charged oligonucleotides they may be recognized by Toll-like receptors (TLRs) and may trigger innate immune reactions. Each of these obstacles has already been separately solved; combinations for overcoming all barriers are being developed. Different approaches are being considered for the development of siRNA and related RNAi modulating therapeutics: chemical and covalent conjugate approaches, which tend to produce small drug-like entities; encapsulation and complexation approaches, which favor nanoparticle formation. The debate remains open whether RNAi compounds should be designed to be as small molecules as possible, to be able to diffuse to and into the target cell almost like a standard drug, or whether siRNA molecules should be provided as cohorts of multiple copies packaged into a larger nanoparticle.2.1 The chemistry approach
A more than thirty years experience in antisense oligonucleotide chemistry has been utilized to make RNAi oligonucleotides metabolically much more stable and non-immunogenic.14,15 For example (see Fig. 2), substituting the natural ribonucleotides by 2′-O-methyl or 2′-F ribonucleotides and introducing phosphorothioates into the oligonucleotide backbones enhances stability and reduces TLR activation. | ||
| Fig. 2 Structures of oligonucleotide modifications. PS, stabilizing phosphorothioate linkages; 2′-OMe, 2′-O-methyl-RNA; 2′-F, 2′-fluoro-RNA; MOE, 2′-O-methoxyethoxy-RNA; LNA, locked nucleic acid nucleoside; tc-DNA, tricycle-DNA nucleoside. | ||
Chemistry also addresses the question: can RNAi oligos be minimized into smaller more drug-like molecules? The structural space for chemical modification however is limited by special requirements in the RNA–RNA interactions with Ago within the RISC. In this respect, chemically bridged ribonucleotides with their conformation locked in A-form have been assembled into locked nucleic acids (LNAs). Due to strong RNA base pairing, LNAs are very effective as antagomirs; even eight nucleotides short “tiny LNAs” which target the seed-region are sufficient for microRNA inactivation.16
Shortening of oligonucleotides such as LNAs to 16 or less nucleotides improves intracellular delivery.17 Endocytic processes named ‘gymnosis’ result in therapeutically relevant cytosolic levels without the help of additional carrier agents. Tricyclo-DNA (tc-DNA) is another new class of RNA-binding oligonucleotides with encouraging pharmacological properties.18
Structural requirements for agonistic siRNAs and microRNAs are more demanding than those for antagonistic antogomirs. For RISC incorporation, the double stranded siRNA has to be delivered. The most recent breakthrough in the field has been the development of potent single stranded siRNA.19,20 Two measures were required for this success: optimizing the chemical stability of single stranded ss-siRNA by introducing alternating 2′-F and 2′-OMe nucleosides in the strand, 2′-methoxyethyl at the ends, and phosphorothioates in most positions. Without these modifications single stranded RNA would be far more vulnerable than double stranded siRNA. The second key measure was the introduction of 5′-vinylphosphonate as metabolically stable mimics of 5′-phosphorylated RNA required in RISC formation. Importantly, stabilized ss-siRNA mediated effective gene silencing also in vivo, illustrated by factor VII and apoCIII mRNA knockdown in the liver upon intravenous administration,19 or suppression of mutant huntingtin in the CNS upon intracerebral spinal fluid administration in a Huntington mouse model.20
2.2 The conjugate approach
Chemical stabilization and size minimization very favourably improve the pharmacological properties of RNAi modulating oligonucleotides, but do not solve the delivery problem completely. A more effective and ideally tissue-targeted intracellular delivery is requested. Thus, targeting ligands21 such as galactoside derivatives,22 folic acid,23 cholesterol or lipids19,24,25 have been conjugated with oligonucleotides or siRNA (see Fig. 3A). Cholesterol-modification mediates lipoprotein binding in blood, thus preventing renal clearance, and triggers cellular uptake by the LDL receptor. Ligand incorporation may support cell binding, possibly also the uptake by endocytosis, however does not resolve the intracellular release problem. | ||
| Fig. 3 Covalent conjugates for RNAi delivery. (A) Monofunctional modifications include receptor targeting ligands such as N-acetyl-galactosamine (NAG), or lipophilic modifications such as cholesterol. (B) An example of a multifunctional dynamic polyconjugate.26 The amide bonds are cleaved at intracellular endosomal acidic pH, the disulfide bond between siRNA and carrier polymer reduced by glutathione (GSH) within the cytoplasm. | ||
Different types of covalent conjugates of siRNA with cationic polymers have been synthesized, with the polycations neutralizing the negative oligonucleotide charges and, after cellular uptake by endocytosis, promoting escape from endosomes into the cytosol.26,27 Such ‘dynamic polyconjugates’ (Fig. 3B) may incorporate several functions (targeting ligands, surface shielding with poly(ethylene glycol) (PEG), endosomolytic domains), however present structures significantly larger than a single siRNA molecule. Smaller and molecularly more precise cationic oligospermine siRNA conjugates were prepared stepwise on an oligonucleotide synthesizer. Such a cationization was sufficient for carrier-free siRNA transfection in cell culture.28
2.3 The encapsulation approach
Different from the small molecule and conjugate approaches, siRNA molecules can be encapsulated in multiple copies into larger structures. Liposomal systems with bioreversible PEG shielding and sizes of around 100 nm are very effective delivery vehicles for various drugs including siRNA. For these purposes SNALPs (stable nucleic acid lipid particles) were developed.29,30 The observed tropism resulting in high gene silencing activity in liver hepatocytes is based on association with lipoprotein apoE during blood circulation as a ligand for receptor-mediated uptake.31 The fusogenicity of incorporated lipids supports the cytosolic release of the multiple incorporated siRNA molecules. It is not surprising that several encouraging clinical therapeutic RNAi studies targeting the liver are based on this robust platform.2.4 The complexation approach
Negatively charged molecules such as oligonucleotides or siRNA can be complexed with recombinant nucleic acid binding proteins,13 peptides,32–34 cationic nature-derived polymers like chitosan35 or synthetic polycations such as various polyethylenimine derivatives36,37 into various nanoparticle forms (‘polyplexes’).38,39 Interestingly, polyplex sizes and stabilities are much more sensitive to the nature of the nucleic acid payload40 as compared to liposomal formulation, where the lipid bilayer but not the encapsulated material determines biophysical properties. This can be regarded as an advantage or as a disadvantage. On the one hand, a polymer formulation optimized for plasmid DNA (pDNA) may not at all be applicable for siRNA. Thus, several strategies have converted small siRNA into larger pDNA-like structures.41,42 On the other hand, tuning of polymers for a given nucleic acid payload can provide nanoparticles with diverse properties, for example with polyplex sizes as small as 7 nm (for polymer-decorated single siRNA molecules) or as large as a micrometer (for siRNA aggregates). Polymers, in contrast to cationic lipids, usually are molecularly less precise, polydisperse molecules. This drawback, however, can be overcome by recent chemical designs such as solid-phase supported assembly of sequence-defined polymers.43–45Fig. 4 displays a sequence-defined ligand-PEG-cationic oligomer used for functional monomeric siRNA polyplex formation.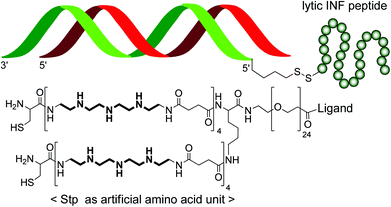 | ||
| Fig. 4 Complexation with polymers. An example of 6 nm small monomolecular siRNA polyplexes formed with sequence-defined oligomers is presented.46 Oligomers contain a targeting ligand (folic acid), a poly(ethylene glycol) (PEG) unit for particle surface shielding, an endosomal protonatable artificial amino acid (Stp), and terminal cysteines for polyplex stabilization by disulfide formation. The siRNA is covalently linked to an endosomolytic peptide. | ||
3. Design in RNAi therapeutics: quo vadis?
The different presented approaches (optimized oligonucleotide chemistry; conjugates; liposomal formulation; polymer-based nanoparticles) synergize in several aspects. Incorporation of functional transport domains, such as shielding in the blood circulation, cell targeting, or endosomal membrane penetration and combinations, can be beneficial in most of the formulation approaches as has already been demonstrated in several cases.23,26,46 For example, receptor-targeted siRNA conjugates can benefit from complexation with endosomally active polymers.23 Chemical oligonucleotide stabilization will be very useful also for all delivery approaches.In other aspects – especially in the size dimension – the strategies go into completely different directions (Fig. 5A). Will minimized small carrier-free oligonucleotide strands be designed exerting still meaningful target sequence specificity? Or will small targeted nanoparticles or larger micro–nano composites be the preferred RNAi drug option?
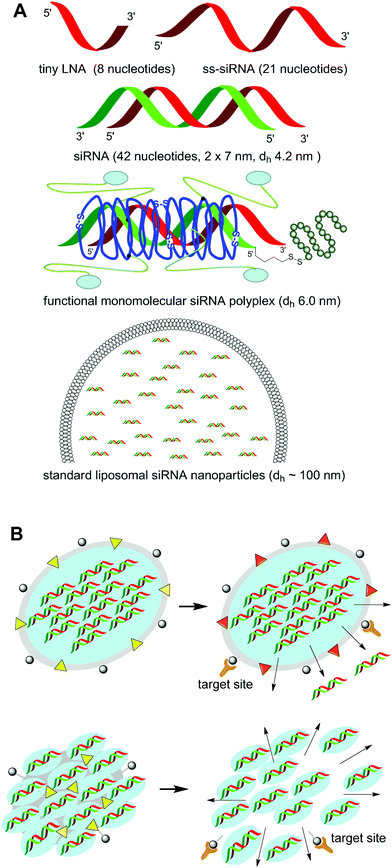 | ||
| Fig. 5 RNAi delivery strategies with diverse characteristics. (A) Different dimensions of RNAi molecules and formulations. (B) Shuttle-type RNAi delivery systems consisting of larger shielded and targeted nanoparticles which deliver their payload to the diseased region, where the specific altered microenvironment is supposed to trigger the release of smaller RNAi subunits. | ||
The answer may depend on the medical indication, the physiology of the affected biological tissue, and the kind of (local, topical or systemic) administration. Furthermore, one larger targeted polyplex, if successfully delivered into a cell, may release an armada of hundreds of siRNA molecules into the cytosol. For the same effect mediated by free or monomolecular complexed siRNA sneaking into the cell, hundreds of successful events would be required. Thus, medium–large nanoparticles and shielded liposomes might be interesting for intravenous targeting of areas with enhanced vasculature permeability such as tumors or inflammation sites, where they benefit from passive retention.47 In contrast, in tissues with restricted drug diffusion, very small RNAi compounds might be advantageous.
Future RNAi therapeutic formulations can be envisioned which combine the best of all the mentioned diverging properties and act like shuttle-based systems (Fig. 5B): larger shielded and targeted nanoparticles (based on lipids, polymers or other controlled release scaffolds) deliver their payload to the diseased region, where biologically triggered smaller drug-loaded subunits are released which facilitate the intracellular transport and subsequent time-controlled extended release of the minimized RNAi drug.
4. Conclusion and prospects
The recent knowledge about RNA interference has yielded powerful tools for manipulating gene expression both for pharmacological scientific and therapeutic purposes. The typical active drug substances present synthetic double stranded siRNAs of 21 base pairs for the sequence-specific recognition and cleavage of mRNA complementary to the siRNA guide strand, but they can be as small as 8 nucleotide tiny LNAs to act as antagomirs or as large as a viral gene vector expressing an RNAi gene. Based on oligonucleotide structure, the novel class of RNAi drugs resides between classical small chemical drugs and macromolecular biological drugs (such as proteins). Current developments have already proven that RNAi drugs can be optimized into biologically stabilized drug-like molecules, which can be synthesized with high precision like conventional drugs. Despite the far more limited intracellular bioavailability as compared with classical drugs, potent RNA interference in vivo has been obtained in some cases. It will be interesting to see whether further chemical tuning of the oligonucleotide chemistry will open up the pharmacological window for broader therapeutic use. Alternatively or in combination, the RNAi drug substance can be incorporated into controlled release formulations. Enhanced delivery to the target tissue, better intracellular uptake, and extended local release may favourably improve both the specificity and pharmacokinetics of the RNAi drug. Improved macromolecular chemistry such as solid-phase-supported syntheses, and innovative nanotechnology assembly methods including microfluidic technologies are available for designing more sophisticated multifunctional, but chemically still precise carriers. It may depend on the route of administration and the disease indication whether the road of optimization will lead to drug-like single RNAi molecule conjugate ‘nanoagents’ or towards larger delivery shuttles locally or intracellularly releasing an armada of RNAi drug molecules.Acknowledgements
The German Research Foundation is gratefully acknowledged for financial support of RNAi related research by the author within the Cluster of Excellence Nanosystems Initiative Munich (NIM) and special research focus project grant SFB1032 B4.References
- A. Fire, S. Xu, M. K. Montgomery, S. A. Kostas, S. E. Driver and C. C. Mello, Nature, 1998, 391, 806–811 CrossRef CAS.
- miRBase: the microRNA database,http://www.mirbase.org.
- Y. Wang, G. Sheng, S. Juranek, T. Tuschl and D. J. Patel, Nature, 2008, 456, 209–213 CrossRef CAS.
- A. Turchinovich and B. Burwinkel, RNA Biol., 2012, 9, 1066–1075 CrossRef CAS.
- S. Polikepahad and D. B. Corry, Nucleic Acids Res., 2013, 41, 1164–1177 CrossRef CAS.
- S. M. Elbashir, J. Harborth, W. Lendeckel, A. Yalcin, K. Weber and T. Tuschl, Nature, 2001, 411, 494–498 CrossRef CAS.
- J. C. Burnett, J. J. Rossi and K. Tiemann, Biotechnol. J., 2011, 6, 1130–1146 CrossRef CAS.
- C. V. Pecot, G. A. Calin, R. L. Coleman, G. Lopez-Berestein and A. K. Sood, Nat. Rev. Cancer, 2011, 11, 59–67 CrossRef CAS.
- D. Haussecker, Mol. Ther.–Nucleic Acids, 2012, 1, e8 CrossRef.
- J. Kota, R. R. Chivukula, K. A. O'Donnell, E. A. Wentzel, C. L. Montgomery, H. W. Hwang, T. C. Chang, P. Vivekanandan, M. Torbenson, K. R. Clark, J. R. Mendell and J. T. Mendell, Cell, 2009, 137, 1005–1017 CrossRef CAS.
- A. F. Ibrahim, U. Weirauch, M. Thomas, A. Grunweller, R. K. Hartmann and A. Aigner, Cancer Res., 2011, 71, 5214–5224 CrossRef CAS.
- J. Krützfeldt, N. Rajewsky, R. Braich, K. G. Rajeev, T. Tuschl, M. Manoharan and M. Stoffel, Nature, 2005, 438, 685–689 CrossRef.
- B. R. Meade and S. F. Dowdy, Discov. Med., 2009, 8, 253–256 Search PubMed.
- M. A. Behlke, Oligonucleotides, 2008, 18, 305–319 CrossRef CAS.
- S. Shukla, C. S. Sumaria and P. I. Pradeepkumar, ChemMedChem, 2010, 5, 328–349 CrossRef CAS.
- S. Obad, C. O. dos Santos, A. Petri, M. Heidenblad, O. Broom, C. Ruse, C. Fu, M. Lindow, J. Stenvang, E. M. Straarup, H. F. Hansen, T. Koch, D. Pappin, G. J. Hannon and S. Kauppinen, Nat. Genet., 2011, 43, 371–380 CrossRef CAS.
- N. Souleimanian, G. F. Deleavey, H. Soifer, S. Wang, K. Tiemann, M. J. Damha and C. A. Stein, Mol. Ther.–Nucleic Acids, 2012, 1, e43 Search PubMed.
- S. Murray, D. Ittig, E. Koller, A. Berdeja, A. Chappell, T. P. Prakash, M. Norrbom, E. E. Swayze, C. J. Leumann and P. P. Seth, Nucleic Acids Res., 2012, 40, 6135–6143 CrossRef CAS.
- W. F. Lima, T. P. Prakash, H. M. Murray, G. A. Kinberger, W. Li, A. E. Chappell, C. S. Li, S. F. Murray, H. Gaus, P. P. Seth, E. E. Swayze and S. T. Crooke, Cell, 2012, 150, 883–894 CrossRef CAS.
- D. Yu, H. Pendergraff, J. Liu, H. B. Kordasiewicz, D. W. Cleveland, E. E. Swayze, W. F. Lima, S. T. Crooke, T. P. Prakash and D. R. Corey, Cell, 2012, 150, 895–908 CrossRef CAS.
- M. Ogris and E. Wagner, Hum. Gene Ther., 2011, 22, 799–807 CrossRef CAS.
- M. Oishi, Y. Nagasaki, K. Itaka, N. Nishiyama and K. Kataoka, J. Am. Chem. Soc., 2005, 127, 1624–1625 CrossRef CAS.
- C. Dohmen, T. Frohlich, U. Lachelt, I. Rohl, H.-P. Vornlocher, P. Hadwiger and E. Wagner, Mol. Ther.–Nucleic Acids, 2012, 1, e7 CrossRef.
- J. Soutschek, A. Akinc, B. Bramlage, K. Charisse, R. Constien, M. Donoghue, S. Elbashir, A. Geick, P. Hadwiger, J. Harborth, M. John, V. Kesavan, G. Lavine, R. K. Pandey, T. Racie, K. G. Rajeev, I. Rohl, I. Toudjarska, G. Wang, S. Wuschko, D. Bumcrot, V. Koteliansky, S. Limmer, M. Manoharan and H. P. Vornlocher, Nature, 2004, 432, 173–178 CrossRef CAS.
- B. Oberhauser and E. Wagner, Nucleic Acids Res., 1992, 20, 533–538 CrossRef CAS.
- D. B. Rozema, D. L. Lewis, D. H. Wakefield, S. C. Wong, J. J. Klein, P. L. Roesch, S. L. Bertin, T. W. Reppen, Q. Chu, A. V. Blokhin, J. E. Hagstrom and J. A. Wolff, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12982–12987 CrossRef CAS.
- M. Meyer, C. Dohmen, A. Philipp, D. Kiener, G. Maiwald, C. Scheu, M. Ogris and E. Wagner, Mol. Pharmaceutics, 2009, 6, 752–762 CrossRef CAS.
- M. Nothisen, M. Kotera, E. Voirin, J. S. Remy and J. P. Behr, J. Am. Chem. Soc., 2009, 131, 17730–1 CrossRef CAS.
- T. S. Zimmermann, A. C. Lee, A. Akinc, B. Bramlage, D. Bumcrot, M. N. Fedoruk, J. Harborth, J. A. Heyes, L. B. Jeffs, M. John, A. D. Judge, K. Lam, K. McClintock, L. V. Nechev, L. R. Palmer, T. Racie, I. Rohl, S. Seiffert, S. Shanmugam, V. Sood, J. Soutschek, I. Toudjarska, A. J. Wheat, E. Yaworski, W. Zedalis, V. Koteliansky, M. Manoharan, H. P. Vornlocher and I. MacLachlan, Nature, 2006, 441, 111–114 CrossRef CAS.
- S. C. Semple, A. Akinc, J. Chen, A. P. Sandhu, B. L. Mui, C. K. Cho, D. W. Y. Sah, D. Stebbing, E. J. Crosley, E. Yaworski, I. M. Hafez, J. R. Dorkin, J. Qin, K. Lam, K. G. Rajeev, K. F. Wong, L. B. Jeffs, L. Nechev, M. L. Eisenhardt, M. Jayaraman, M. Kazem, M. A. Maier, M. Srinivasulu, M. J. Weinstein, Q. Chen, R. Alvarez, S. A. Barros, S. De, S. K. Klimuk, T. Borland, V. Kosovrasti, W. L. Cantley, Y. K. Tam, M. Manoharan, M. A. Ciufolini, M. A. Tracy, A. de Fougerolles, I. MacLachlan, P. R. Cullis, T. D. Madden and M. J. Hope, Nat. Biotechnol., 2010, 28, 172–176 CrossRef CAS.
- A. Akinc, W. Querbes, S. De, J. Qin, M. Frank-Kamenetsky, K. N. Jayaprakash, M. Jayaraman, K. G. Rajeev, W. L. Cantley, J. R. Dorkin, J. S. Butler, L. Qin, T. Racie, A. Sprague, E. Fava, A. Zeigerer, M. J. Hope, M. Zerial, D. W. Sah, K. Fitzgerald, M. A. Tracy, M. Manoharan, V. Koteliansky, A. Fougerolles and M. A. Maier, Mol. Ther., 2010, 18, 1357–1364 CrossRef CAS.
- T. Frohlich and E. Wagner, Soft Matter, 2010, 6, 226–234 RSC.
- S. E. Andaloussi, T. Lehto, I. Mager, K. Rosenthal-Aizman, I. I. Oprea, O. E. Simonson, H. Sork, K. Ezzat, D. M. Copolovici, K. Kurrikoff, J. R. Viola, E. M. Zaghloul, R. Sillard, H. J. Johansson, F. Said Hassane, P. Guterstam, J. Suhorutsenko, P. M. Moreno, N. Oskolkov, J. Halldin, U. Tedebark, A. Metspalu, B. Lebleu, J. Lehtio, C. I. Smith and U. Langel, Nucleic Acids Res., 2011, 39, 3972–3987 CrossRef.
- L. Crombez and G. Divita, Methods Mol. Biol., 2011, 683, 349–360 CAS.
- K. A. Howard, U. L. Rahbek, X. Liu, C. K. Damgaard, S. Z. Glud, M. O. Andersen, M. B. Hovgaard, A. Schmitz, J. R. Nyengaard, F. Besenbacher and J. Kjems, Mol. Ther., 2006, 14, 476–484 CrossRef CAS.
- G. Creusat, J. S. Thomann, A. Maglott, B. Pons, M. Dontenwill, E. Guerin, B. Frisch and G. Zuber, J. Controlled Release, 2012, 157, 418–426 CrossRef CAS.
- A. Zintchenko, A. Philipp, A. Dehshahri and E. Wagner, Bioconjugate Chem., 2008, 19, 1448–1455 CrossRef CAS.
- E. Wagner, Acc. Chem. Res., 2012, 45, 1005–1013 CrossRef CAS.
- K. Miyata, N. Nishiyama and K. Kataoka, Chem. Soc. Rev., 2012, 41, 2562–2574 RSC.
- C. Scholz and E. Wagner, J. Controlled Release, 2012, 161, 554–565 CrossRef CAS.
- A. L. Bolcato-Bellemin, M. E. Bonnet, G. Creusat, P. Erbacher and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16050–16055 CrossRef CAS.
- S. Y. Lee, M. S. Huh, S. Lee, S. J. Lee, H. Chung, J. H. Park, Y. K. Oh, K. Choi, K. Kim and I. C. Kwon, J. Controlled Release, 2010, 141, 339–346 CrossRef CAS.
- D. Schaffert, C. Troiber, E. E. Salcher, T. Frohlich, I. Martin, N. Badgujar, C. Dohmen, D. Edinger, R. Klager, G. Maiwald, K. Farkasova, S. Seeber, K. Jahn-Hofmann, P. Hadwiger and E. Wagner, Angew. Chem., Int. Ed., 2011, 50, 8986–8989 CrossRef CAS.
- D. Schaffert, N. Badgujar and E. Wagner, Org. Lett., 2011, 13, 1586–1589 CrossRef CAS.
- T. Frohlich, D. Edinger, R. Klager, C. Troiber, E. Salcher, N. Badgujar, I. Martin, D. Schaffert, A. Cengizeroglu, P. Hadwiger, H. P. Vornlocher and E. Wagner, J. Controlled Release, 2012, 160, 532–541 CrossRef.
- C. Dohmen, D. Edinger, T. Frohlich, L. Schreiner, U. Lachelt, C. Troiber, J. Radler, P. Hadwiger, H. P. Vornlocher and E. Wagner, ACS Nano, 2012, 6, 5198–5208 CrossRef CAS.
- H. Maeda, Adv. Enzyme Regul., 2001, 41, 189–207 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |