Sugar/gadolinium-loaded gold nanoparticles for labelling and imaging cells by magnetic resonance imaging†
Ainhoa
Irure
a,
Marco
Marradi
ab,
Blanca
Arnáiz
a,
Nuria
Genicio
a,
Daniel
Padro
c and
Soledad
Penadés
*ab
aLaboratory of Glyconanotechnology, Biofunctional Nanomaterials Unit, CIC biomaGUNE, P° Miramón 182, 20009 San Sebastián, Spain. E-mail: spenades@cicbiomagune.es; Fax: +34 943 00 53 35; Tel: +34 943 00 53 07
bBiomedical Research Networking Center in Bioengineering, Biomaterials and Nanomedicine (CIBER-BBN), P° Miramón 182, 20009 San Sebastián, Spain
cMolecular Imaging Unit, CIC biomaGUNE, P° de Miramón 182, 20009 San Sebastián, Spain
First published on 4th April 2013
Abstract
Targeted magnetic resonance imaging (MRI) probes for selective cell labelling and tracking are highly desired. We here present biocompatible sugar-coated paramagnetic Gd-based gold nanoparticles (Gd-GNPs) and test them as MRI T1 reporters in different cellular lines at a high magnetic field (11.7 T). With an average number of 20 Gd atoms per nanoparticle, Gd-GNPs show relaxivity values r1 ranging from 7 to 18 mM−1 s−1 at 1.41 T. The multivalent presentation of carbohydrates on the Gd-GNPs enhances the avidity of sugars for carbohydrate-binding receptors at the cell surface and increases the local concentration of the probes. A large reduction in longitudinal relaxation times T1 is achieved with both fixed cells and live cells. Differences in cellular labelling are obtained by changing the type of sugar on the gold surface, indicating that simple monosaccharides and disaccharides are able to modulate the cellular uptake. These results stress the benefits of using sugars to produce nanoparticles for cellular labelling. To the best of our knowledge this is the first report on labelling and imaging cells with Gd-based gold nanoparticles which use simple sugars as receptor reporters.
1. Introduction
The in vivo targeting of nanoparticles to a specific tissue or organ is of great interest for diagnostic and therapeutic purposes, but encounters several problems such as selectivity, biodistribution, and toxicity.1–4 The application of nanoparticles in biomedicine requires a much deeper understanding of the in vivo trafficking and fate of the nanoconjugates. Molecular imaging technologies such as magnetic resonance imaging (MRI), X-ray computed tomography (CT), positron emission tomography (PET), single-photon emission computed tomography (SPECT), and ultrasound (US) are essential for this development. Among these techniques, non-invasive MRI is preferred for cell imaging because of its better resolution.5 The relatively low sensitivity of MRI can be improved by the use of contrast agents, which produces a hyperintense (white) or hypointense (black) signal by shortening the T1 (paramagnetic) and T2 (superparamagnetic) proton relaxation times.6 Iron oxide nanoparticles are the most widely used T2-contrast agents for cell imaging. However, paramagnetic gadolinium(III) complexes are also promising in cell tracking and fate mapping experiments.7 Because of the low intracellular labelling efficiency and the quenching of relaxivity once internalized, examples of cell labelling with T1 gadolinium-based contrast agents are limited.5 To overcome these shortcomings, strategies have been developed for increasing the intracellular delivery of nanoparticles focused on small molecule contrast agents that report on enzyme activity,8–10 target proteins,11 or enhance cell penetration.12–14 Other approaches include the use of a variety of scaffolds such as Gd-loaded liposomes or micelles,15,16 polymers,17 dendrimers,18–20 and nanoparticles.21,22Gold nanoparticles are also appealing scaffolds due to the opportunity for easy conjugation of Gd chelates on the metal surface23–25 in combination with other (bio)molecules.26–31 By attaching thiol-derivatized Gd(III) chelates on gold nanoclusters, relaxivities ranging from 3 to 60 mM−1 s−1 have been obtained.25 Lim et al.28 labelled cancer cells with hollow gold nanoparticles functionalized with anti-HER2 antibodies chemically conjugated to DTPA-Gd. Song et al.29 prepared Gd-enriched poly-dT oligonucleotides gold nanoparticles that could penetrate NIH/3T3 cells to allow successful cellular MR imaging at high fields. Peptides, proteins, and antibodies are the most frequently used molecules to obtain targeted nanoparticles and contrast agents.3,4,32,33 Antibodies are the preferential vectors to accumulate contrast agents in cells that express the targeted markers because of their highly specific recognition properties.16,28 On the other hand, less attention has been paid to carbohydrates as targeting molecules.
Carbohydrates are, together with proteins and nucleic acids, the third group of significant biological molecules. They have great structural diversity ranging from single monosaccharides to complex oligosaccharides and polysaccharides (glycans) and contain a wealth of information for triggering cellular response.34–36 Simple sugars are important nutrients for cell growth, while complex glycans (glycoproteins, glycolipids) are specific signalling molecules. The structural diversity and their role in many cellular events make carbohydrates attractive molecules for developing targeted contrast agents. Furthermore, carbohydrates are highly hydrophilic, ensuring both water solubility and biocompatibility to the imaging probe and avoiding clearance in vivo by macrophages.37
The potential of protein–sugar interaction for specific delivery of therapeutic drugs was appreciated early.38 Although nanoparticles coated with carbohydrates have been employed for the studies of carbohydrate-mediated interactions39 few examples report on sugar-based targeted magnetic nanoparticles. A recent review highlighted the use of glycans in the preparation of iron oxide and Gd probes for MRI.40 Among superparamagnetic iron oxide MRI T2-probes, Huang and co-workers recently reported on nanoparticles coated with simple sugars that are able to target and differentiate cancer cells.41
Carbohydrates bind to their cellular receptors (lectins)42 with low affinity, but the avidity for the receptors highly increases when carbohydrates are presented in a multivalent fashion (cluster effect).43 Glyconanoparticles (GNPs) are carbohydrate-coated gold nanoparticles that mimic the multivalent presentation of glycans at the cell surfaces.44,45 These GNPs were originally prepared to investigate carbohydrate-mediated interactions,46 but more recently, based on the high glucose uptake typical of tumour derived cells (the Warburg effect),47 we prepared the first gold nanoparticles coated with multiple copies of sugars and Gd-complexes (Gd-GNPs) as T1-agents aiming at targeting glioma in mouse brain.48 We observed that Gd-GNP coated with glucose conjugates enhanced in vivo the MRI contrast in murine glioma better than Magnevist™ (a T1-contrast agent in clinical use), while Gd-GNP bearing the disaccharide lactose was not able to reach the tumour. As the nature of the sugar seems to influence the in vivo behaviour of Gd-GNPs, we decided to prepare a series of sugar-coated Gd-GNPs and test them as T1 receptor reporters in cellular models. The differential transport and uptake of simple sugars (glucose, galactose, mannose, glucosamine) by cells depend on the expression of GLUT transporters49 and sugar receptors (lectins), which could help to achieve cell permeable MRI probes and in vitro cell selectivity.
In this work, we have explored the selectivity of simple carbohydrates for targeting sugar cell transporters and lectins and the potential of MRI as an in vitro screening method for investigating the interactions between nanoparticles and cells.50,51 We expected that the combination of multiple glycoconjugates and paramagnetic complexes on the very same gold nanocluster should help to target cellular surface receptors with high avidity. Therefore, we have prepared a small library of Gd-based glyconanoparticles (Gd-GNPs), incorporating multiple copies of Gd(III)-complexes and simple carbohydrates, with well-defined composition and optimized relaxivity values. Some of them were selected for investigating in vitro their interaction with four different cellular lines at a high magnetic field (11.7 T).
2. Experimental methods
2.1 General methods and materials
All chemicals were purchased as reagent grade from Sigma-Aldrich and were used without further purification. UV-Vis spectra were carried out with a Beckman Coulter DU 800 spectrometer. Infrared spectra (IR) were recorded from 4000 to 500 cm−1 with a JASCO FT/IR 410 model spectrometer: solids were pressed into a KBr plate and oils were subjected to attenuated total reflection (ATR). 1H NMR and 13C NMR spectra were recorded using a Bruker AVANCE (500 MHz) spectrometer. Chemical shifts (δ) are given in ppm relative to the residual signal of the solvent used. Coupling constants (J) are reported in Hz. Splitting patterns are described by using the following abbreviations: br, broad; s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet. Mass spectra were carried out with an Esquire 6000 ESIIon Trap from Bruker Daltonics. High resolution mass spectra (HR-MS) were obtained using the matrix-assisted laser desorption/ionization (MALDI) technique with a 4700 Proteomics Analyzer (Applied Biosystems) with MALDI-time-of-flight (TOF) configuration. For transmission electron microscopy (TEM) examinations, a single drop (1 μL) of the aqueous solution (ca. 0.1 mg mL−1 in MilliQ water) of the gold glyconanoparticles (GNPs) was placed onto a copper grid coated with a carbon film (Electron Microscopy Sciences). The grid was left to dry in air for several hours at room temperature. TEM analysis was carried out in a Philips JEOL JEM-2100F working at 200 kV. The average diameter and the number of gold atoms of the GNPs were deduced according to a previous work.52 House distilled water was further purified using a Milli-Q reagent grade water system (Millipore).2.2 Incubation of SCxDO3A (x = 5 or 11) derivatives with Gd(III)
To chelate the Gd(III) cations, 1 eq. of a DO3A derivative (SC5DO3A and SC11DO3A), prepared as reported in the ESI,† was dissolved in 1.5 mL of HEPES buffer (0.1 M, pH 7.4) and then incubated with 0.9 equivalents of an aqueous solution of GdCl3 (0.5 mL, 0.1 M) at 25 °C for 2 hours. The freshly prepared HEPES solutions were then used for the ligand place exchange reaction (LPE) with 100% sugar nanoparticles.2.3 Preparation of sugar/Gd-coated gold nanoparticles (Gd-GNPs) by ligand place exchange (LPE) reaction
100% Sugar-coated gold glyconanoparticles (1 eq. of the sugar), prepared as reported in the ESI,† were dissolved in MilliQ water and a HEPES solution of the previously prepared SCxDO3A-Gd complex (1.1 eq. with respect to the sugar coating) was added. The mixture was stirred in an orbital shaker for 44 hours at 25 °C and 180 rpm. The solution was filtered with Amicon filters (MWCO = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) until the washing did not show SCxDO3A-Gd. The so-obtained Gd-GNPs were diluted in MilliQ water and freeze-dried.
000 g mol−1) until the washing did not show SCxDO3A-Gd. The so-obtained Gd-GNPs were diluted in MilliQ water and freeze-dried.
2.4 T 1 measurements at 1.41 Tesla
Water proton NMR T1 relaxation times at 1.41 T (60 MHz) were measured at 37 °C in a Bruker Minispec MQ60 TD-NMR spectrometer (Bruker Biospin, Ettlingen, Germany) using serial dilutions of Gd-GNPs (10, 5, 2.5, 1.25, 0.625 mg mL−1) in MilliQ water. The values of T1 were determined using the inversion-recovery method. 300 μL of the solutions were used for each determination and the fitting of the T1 was done using the appropriate inversion–recovery curve. Three different measurements of T1 were performed in every sample for statistical correction. The commercial MRI contrast agent Dotarem® was used as a reference.2.5 T 1 measurements at 11.7 Tesla
Water proton NMR relaxation times T1 at 11.7 T (500 MHz) were measured at 25 °C in a Bruker Biospec using a 72 mm volumetric quadrature coil using serial dilutions of Gd-GNPs at 200, 100, 50, and 5 μM Gd(III) concentrations. Saturation recovery pulse sequences with static TE (11 ms) and variable TR (300, 650, 730, 1100, 1500, 2100, 2800, 3800, 5500, 12500 ms) values were used. Imaging parameters were as follows: field of view (FOV) = 34 × 34 mm2, matrix size (MTX) = 320 × 320, slice thickness 0.5 mm, and 4 or 8 averages. T1 analysis was carried out using the image sequence analysis tool in Paravision 5 software (Bruker BioSpin, Ettlingen, Germany) with mono-exponential curve-fitting of the image intensities of selected regions of interest (ROIs). The corresponding relaxivity values r1 at 1.4 and 11.7 T were calculated from the slopes of the curves of the concentration of Gd(III) expressed in mM vs. 1/T1 (s−1) (ESI, Fig. S3 and S3bis;† for phantoms, see Fig. S4†).
2.6 In vitro cytotoxicity studies
Three different cell lines, namely GL261 (murine glioma cells), HepG2 (hepatocytes) and Raji cells (Burkitt lymphoma cells), were used in this study. The viability of GL261 and HepG2 cells was determined by using the MTT method. Briefly, 104 cells per well were seeded into 96-well plates in a 100 μL complete medium and incubated at 37 °C in a 5% CO2 atmosphere. After 24 hours, the medium was replaced with a fresh one containing nanoparticles at different concentrations (0–100 μg mL−1). After 20 hours incubation period, 20 μL of MTT (5 mg mL−1 in phosphate buffer pH 7.4) was added to each well. After 4 hours of incubation at 37 °C and 5% CO2 for exponentially growing cells and 15 min for steady-state confluent cells, the medium was removed, formazan crystals were dissolved with 200 μL of DMSO, and the solution was vigorously mixed to dissolve the reacted dye. The absorbance of each well was read on a multiplate reader (GENios Pro instrument from TECAN) at 550 nm. The toxicity of the GNPs towards Raji cells was studied in the 0–100 μg mL−1 concentration range using an MTS standard protocol. 1 × 104 cells per well were seeded into 96-well plates in 80 μL of complete medium and then a GNPs solution (20 μL) at a desired concentration was added and incubated at 37 °C in a 5% CO2 atmosphere. After 20 h, 20 μL of an MTS solution (5 mg mL−1) were added to each well and cells were further incubated for 4 h at 37 °C in a 5% CO2 atmosphere. Finally, the absorbance of the samples was measured at 490 nm on the multiplate reader. The standard deviations (±SD) were obtained on a triplicate analysis (n = 3). The relative cell viability for GL261, HepG2, and Raji cells after incubation for 24 h at concentrations up to 100 μg mL−1 of Gd-GNPs is reported in the ESI (Fig. S5†).2.7 MRI phantoms
T 1 measurements, at different concentrations of Gd(III) (0, 5, 50, 100 and 200 μM), were performed in a Bruker Biospec at 11.7 T using a 72 mm volumetric quadrature coil at room temperature. Saturation recovery pulse sequences with static TE (11 ms) and variable TR (300, 650, 730, 1100, 1500, 2100, 2800, 3800, 5500, 12500 ms) values were used. Imaging parameters were as follows: field of view (FOV) = 34 × 34 mm2, matrix size (MTX) = 320 × 320, slice thickness 0.5 mm, and four averages. T1 analysis was carried out using the image sequence analysis tool in Paravision 5 software (Bruker BioSpin, Ettlingen, Germany) with monoexponential curve-fitting of the image intensities of selected regions of interest (ROIs). T1-weighted images were acquired using a Gradient Echo Sequence with 400 ms repetition time and 4.7 ms echo time. Imaging parameters were as follows: field of view (FOV) = 34 × 34 mm2, matrix size (MTX) = 320 × 320, slice thickness 0.5 mm, and four averages (total acquisition time 1 h).
2.8 Determination of Gd-GNPs uptake in fixed cells by MR imaging
GL261, HepG2, Raji and Raji-DC-SIGN were grown in 75 cc tissue culture flasks at 37 °C in an atmosphere of 5% CO2 and 95% air, washed with 10 mM PBS without Ca2+/Mg2+, detached by 10 min incubation at 37 °C with 8 mL of a cell dissociation solution (Sigma-Aldrich) to avoid digestion of cell surface proteins. Cells were then pelleted, re-suspended in 1 mL of 10 mM PBS, disaggregated by passing them through a 25 gauge-needle, counted and seeded in a round bottom 96-well plate (∼0.5–3 × 106 cells per well). Cells were then washed by centrifugation (500g, 5 min, 4 °C) once with warm (37 °C) 10 mM PBS, fixed with 2.5% formaldehyde (10 min at r.t.) and again washed three times with 10 mM PBS to remove any residual formaldehyde. GL261, HepG2, Raji and Raji-DC-SIGN pellets were re-suspended in a 50 μM or 130 μM (Gd(III) concentration) culture media solution of Gd-GNPs or Dotarem® and incubated for 1 hour at 37 °C in an atmosphere of 5% CO2 and 95% air. After incubation, the plate was centrifuged (500g, 5 min, 4 °C) and the supernatant was collected. Pellets were washed three times with cold PBS (10 mM) by centrifugation (500g, 5 min, 4 °C) to remove the unbound Gd-GNPs or Dotarem®. Finally, pellets were re-suspended in ∼30 μL media and transferred into sealed jet Pasteur pipettes. The pipettes were kept at 4 °C overnight to allow pellet formation. The pipettes were cut, placed in a plastic cup, filled with agarose (1%) and then subjected to MRI imaging (11.7 T). GL261 cells were also incubated with GlcC5S-Au-SC11DO3A-Gd (Glc-GNP) at 1.3, 5, 13 or 50 μM concentrations of gadolinium.2.9 Determination of Gd-GNPs uptake in cells alive by MR imaging
GL261 and HepG2 cells were seeded in 6-well plates at 2 × 105 and 2.5 × 105 cells per well, respectively, and grown for 3 days at 37 °C in an atmosphere of 5% CO2 and 95% air. Raji or Raji-DC-SIGN were counted, seeded (∼3 × 106 cells per well) in a round bottom 96-well plate and washed three times with PBS (10 mM) by centrifugation (500g, 10 min, 4 °C). Raji and Raji-DC-SIGN pellets, and GL261 and HepG2 monolayers were incubated for 4 hours in a 50 or 130 μM (Gd(III) concentration) culture media solution of Gd-GNPs or Dotarem® at 37 °C in an atmosphere of 5% CO2 and 95% air. After incubation, supernatants were collected and the cells were washed three times with warm 10 mM PBS to remove the unbound Gd-GNPs or Dotarem®. GL261 and HepG2 cell monolayers were detached from the 6-well plates by trypsination (10 min, at r.t.), transferred to 1.5 mL tubes and pelleted by centrifugation. GL261, HepG2, Raji and Raji-DC-SIGN pellets were re-suspended in ∼30 μL of cold 10 mM PBS and transferred into sealed jet Pasteur pipettes. The pipettes were kept at 4 °C overnight to allow pellet formation. The pipettes were cut, placed in a plastic cup, filled with agarose (1%) and then subjected to MRI imaging (11.7 T).2.10 Cell lysates preparation for MR imaging
Dried pellets of the live HepG2, Raji and Raji-DC-SIGN cells, incubated with Gd-GNPs at 50 μM Gd concentration and transferred to sealed jet Pasteur pipettes, were re-hydrated in 20 μL of a hypotonic buffer (HEPES, 25 mM). After a 1 h incubation at r.t. for hypotonic shock, the remaining whole cells were lysed by 3 freeze–thaw cycles followed by sonication (3 cycles of 15 s; water bath Ultrasons-h, Selecta). The pipettes were placed in a plastic cup, filled with agarose (1%) and then subjected to MRI imaging (11.7 T).3. Results and discussion
3.1 Preparation and properties of Gd-GNPs
The Gd-GNPs were prepared by ligand place exchange reaction (LPE)53 starting from glyconanoparticles bearing only sugars. Gold glyconanoparticles (GNPs) 100% coated with glycoconjugates of the monosaccharides glucose (GlcCnS), galactose (GalCnS), mannose (ManCnS), and the disaccharides lactose (LacCnS), maltose (maltoseCnS), and cellobiose (cellobioseCnS) were prepared as described54 by in situ reduction of a gold salt (HAuCl4) with NaBH4 in the presence of glycoconjugates (Scheme 1). After purification by dialysis and lyophilization, the prepared GNPs were characterized by TEM, UV, 1H NMR, and elemental analysis. The average diameter of the gold core was 1.5–2.5 nm and the number of sugars on the surface ranged between 70 and 130 molecules (see ESI†).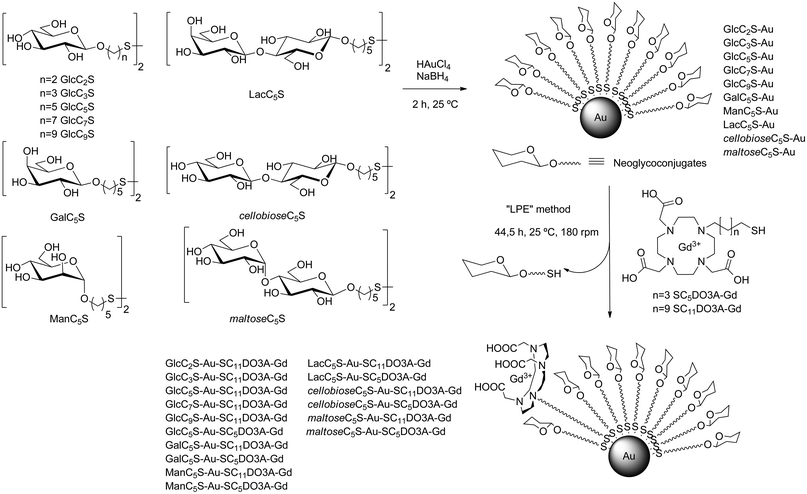 | ||
| Scheme 1 Glycoconjugates used for the synthesis of 100% sugar-coated gold glyconanoparticles (GNPs) and incorporation of DO3A-Gd complexes by ligand place exchange reaction (LPE) to obtain the sugar/Gd-coated gold nanoparticles (Gd-GNPs). | ||
The GNPs were converted into the Gd-based gold glyconanoparticles (Gd-GNPs) by LPE reaction with thiol-ending functionalized 1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid (DO3A) Gd complexes (SC5DO3A-Gd or SC11DO3A-Gd) (Scheme 1, and ESI Table S1†).
The glycoconjugates of β-glucose (GlcC2S, GlcC3S, GlcC5S, GlcC7S, and GlcC9S), β-galactose (GalC5S), and α-mannose (ManC5S) were prepared as already described.55,56 β-Lactose (LacC5S), β-maltose (maltoseC5S), and β-cellobiose (cellobioseC5S) conjugates were also synthesized following established procedures.54 In order to attach the chelating agent to the gold surface, DO3A was functionalized with a five or eleven carbon atoms aliphatic linker ending in a thiol group to obtain the derivatives SC5DO3A and SC11DO3A (Scheme 1).
Briefly, alkylation of commercial DO3A-tBu with 11-bromo-1-(S-trityl) mercaptoundecane or 5-bromo-1-(S-trityl) mercaptopentane and subsequent deprotection of both t-butyl esters and trityl group in acidic conditions afforded the desired DO3A derivatives (Scheme 2). For the chelating of Gd(III) cations, the DO3A derivatives in HEPES buffer (0.1 M pH 7.4) were incubated with 0.9 equivalents of a water solution of GdCl3 (0.1 M) and the resulting complexes were characterized by IR, inductively coupled plasma atomic emission spectroscopy (ICP-AES) analysis, 17O NMR and relaxivity values (see ESI†).
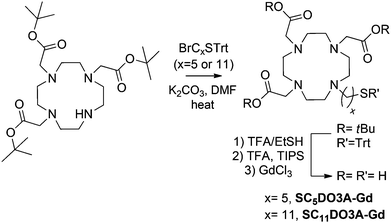 | ||
| Scheme 2 Synthesis of the SCxDO3A derivatives. | ||
To prepare the Gd-GNPs (Scheme 1), a solution of the DO3A-Gd derivatives in HEPES buffer (1.1 eq. with respect to the carbohydrate loading of each GNP) was added to an aqueous solution of the GNPs and incubated at 25 °C for 44 hours and 180 rpm. The solution was filtered by Amicon (MWCO 10000) and then washed several times with water until Gd was not detected in the washings. The obtained Gd-GNPs were characterized by TEM, UV-Vis, IR, 17O NMR, dynamic light scattering (DLS) and ICP-AES (see ESI†). The size-distribution range of the gold core was 1.5–2.5 nm. Comparing the gold nanocluster size before and after the LPE reaction, no significant change in the core size (TEM) and hydrodynamic diameter (DLS) was observed. IR spectra of the Gd-GNPs showed a band at 1600 cm−1 which corresponds to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of COO− coordinated to Gd3+. This band was absent in the starting 100%-sugar-coated gold GNPs and appeared after the LPE, indicating the incorporation of the chelates. The amount of Gd(III) on the GNPs was between 2% and 5% (Table 1) as determined by ICP-AES, and the average number of Gd per nanoparticle between 10 and 30 atoms was calculated based on the gold core sizes as determined by TEM (ESI, Table S1†). Attempts to increase the Gd loading on the GNPs resulted in loss of stability and precipitation of Gd-GNPs. The longitudinal T1 relaxation and transverse T2 times of Gd-GNPs were determined at 10, 5, 2.5, 1.25, 0.625 mg mL−1 concentrations of Gd-GNPs in water at 60 MHz (1.41 T) and 37 °C. The relaxivity values r1 and r2 were obtained from the slopes of the curves, the concentration of Gd(III) expressed in mM vs. 1/T1 or 1/T2 (Table 1). The ratio r2/r1 at 1.41 T is in the range of 1.2–1.9 in agreement with the typical relaxation properties of T1-agents. The relaxation times of Gd-GNPs in water solutions at 200, 100, 50, 5 μM concentrations of Gd were also measured in “phantoms” at 11.7 T and 25 °C (see ESI†). As expected for macromolecules, a dramatic decrease in r1 is observed at high field (Table 1). The relaxivity values at 1.41 T range from 6.3 to 18.0 mM−1 s−1 depending on the molecular structure of glycoconjugates and DO3A-Gd complexes. The Gd-GNPs bearing the SC5-DO3A-Gd complex have higher relaxivity values (11–18 mM−1 s−1) than the ones bearing monosaccharide conjugates and the SC11DO3A-Gd complex (6–9 mM−1 s−1), while those coated with disaccharides and SC11DO3A-Gd have also high r1 values (Table 1). The origin of the r1 increase may be attributed to the different mobility of the Gd complex on the gold surface of the Gd-GNPs (Fig. 1). In the ones bearing monosaccharides and the SC11DO3A-Gd chelate (Fig. 1a), the complex protrudes above the sugar shell, while in those bearing disaccharides (Fig. 1c), as well as monosaccharides with the SC5DO3A-Gd chelate (Fig. 1b), the Gd complexes have similar length to the sugar conjugates. This arrangement produces a packaging effect of the glycoconjugates towards the Gd-chelates, reducing their mobility. The increasing rigidity of the chelate may be responsible for the improvement of the relaxivity values in those Gd-GNPs.
O stretching of COO− coordinated to Gd3+. This band was absent in the starting 100%-sugar-coated gold GNPs and appeared after the LPE, indicating the incorporation of the chelates. The amount of Gd(III) on the GNPs was between 2% and 5% (Table 1) as determined by ICP-AES, and the average number of Gd per nanoparticle between 10 and 30 atoms was calculated based on the gold core sizes as determined by TEM (ESI, Table S1†). Attempts to increase the Gd loading on the GNPs resulted in loss of stability and precipitation of Gd-GNPs. The longitudinal T1 relaxation and transverse T2 times of Gd-GNPs were determined at 10, 5, 2.5, 1.25, 0.625 mg mL−1 concentrations of Gd-GNPs in water at 60 MHz (1.41 T) and 37 °C. The relaxivity values r1 and r2 were obtained from the slopes of the curves, the concentration of Gd(III) expressed in mM vs. 1/T1 or 1/T2 (Table 1). The ratio r2/r1 at 1.41 T is in the range of 1.2–1.9 in agreement with the typical relaxation properties of T1-agents. The relaxation times of Gd-GNPs in water solutions at 200, 100, 50, 5 μM concentrations of Gd were also measured in “phantoms” at 11.7 T and 25 °C (see ESI†). As expected for macromolecules, a dramatic decrease in r1 is observed at high field (Table 1). The relaxivity values at 1.41 T range from 6.3 to 18.0 mM−1 s−1 depending on the molecular structure of glycoconjugates and DO3A-Gd complexes. The Gd-GNPs bearing the SC5-DO3A-Gd complex have higher relaxivity values (11–18 mM−1 s−1) than the ones bearing monosaccharide conjugates and the SC11DO3A-Gd complex (6–9 mM−1 s−1), while those coated with disaccharides and SC11DO3A-Gd have also high r1 values (Table 1). The origin of the r1 increase may be attributed to the different mobility of the Gd complex on the gold surface of the Gd-GNPs (Fig. 1). In the ones bearing monosaccharides and the SC11DO3A-Gd chelate (Fig. 1a), the complex protrudes above the sugar shell, while in those bearing disaccharides (Fig. 1c), as well as monosaccharides with the SC5DO3A-Gd chelate (Fig. 1b), the Gd complexes have similar length to the sugar conjugates. This arrangement produces a packaging effect of the glycoconjugates towards the Gd-chelates, reducing their mobility. The increasing rigidity of the chelate may be responsible for the improvement of the relaxivity values in those Gd-GNPs.
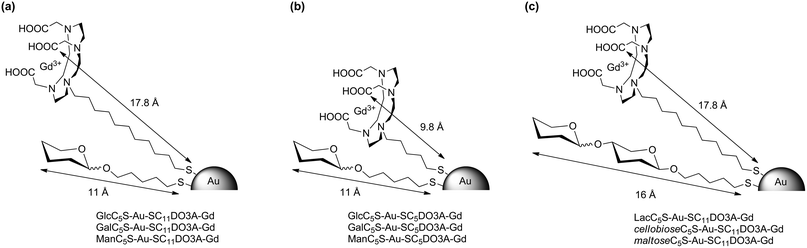 | ||
| Fig. 1 Schematic representation of the paramagnetic Gd-based gold glyconanoparticles (Gd-GNPs) showing the calculated length of the glycoconjugates and the DO3A-Gd complex. | ||
| Gd-GNPsa | TEM (nm) | % Gd | r 1 (mM−1 s−1) 1.41 T | r 2 (s−1 mM−1) 1.41 T | r 1 (mM−1 s−1) 11.7 T |
|---|---|---|---|---|---|
| a In brackets are the Gd-GNPs selected for the cellular assays. | |||||
| GlcC2S-Au-SC11DO3A-Gd | 1.8 ± 0.2 | 3.4 ± 0.2 | 7.1 ± 0.9 | 11.1 ± 0.7 | 1.8 ± 0.8 |
| GlcC3S-Au-SC11DO3A-Gd | 1.9 ± 0.3 | 3.3 ± 0.2 | 6.3 ± 0.7 | 10.5 ± 0.9 | 2.1 ± 0.9 |
| GlcC5S-Au-SC11DO3A-Gd (Glc-GNP) | 1.5 ± 0.3 | 4.7 ± 0.1 | 7.4 ± 0.7 | 11.8 ± 0.8 | 2.5 ± 0.9 |
| GlcC7S-Au-SC11DO3A-Gd | 1.8 ± 0.3 | 4.1 ± 0.2 | 7.1 ± 0.9 | 11.8 ± 0.9 | 2.0 ± 0.7 |
| GlcC9S-Au-SC11DO3A-Gd | 1.8 ± 0.2 | 3.2 ± 0.2 | 7.5 ± 0.6 | 13.0 ± 1.0 | 2.7 ± 0.9 |
| GlcC5S-Au-SC5DO3A-Gd | 2.0 ± 0.3 | 3.1 ± 0.2 | 16.9 ± 0.4 | 27.4 ± 1.6 | 4.0 ± 0.6 |
| GalC5S-Au-SC11DO3A-Gd (Gal-GNP) | 1.7 ± 0.1 | 4.2 ± 0.2 | 8.0 ± 0.9 | 12.9 ± 0.9 | 2.1 ± 0.9 |
| GalC5S-Au-SC5DO3A-Gd | 1.7 ± 0.2 | 3.6 ± 0.2 | 18.0 ± 0.3 | 30.4 ± 1.8 | 4.1 ± 0.8 |
| ManC5S-Au-SC11DO3A-Gd (Man-GNP) | 2.4 ± 0.4 | 3.0 ± 0.2 | 9.7 ± 0.6 | 16.3 ± 1.1 | 2.3 ± 0.6 |
| ManC5S-Au-SC5DO3A-Gd | 1.8 ± 0.2 | 4.1 ± 0.2 | 15.5 ± 0.9 | 25.6 ± 1.7 | 3.8 ± 0.9 |
| LacC5S-Au-SC11DO3A-Gd (Lac-GNP) | 1.7 ± 0.3 | 2.7 ± 0.2 | 12.9 ± 0.9 | 15.7 ± 0.9 | 4.4 ± 0.9 |
| LacC5S-Au-SC5DO3A-Gd | 1.6 ± 0.2 | 3.7 ± 0.2 | 17.0 ± 0.8 | 29.2 ± 1.7 | 3.8 ± 0.7 |
| CellobioseC5S-Au-SC11DO3A-Gd (cellobiose-GNP) | 1.8 ± 0.2 | 2.1 ± 0.2 | 11.2 ± 0.9 | 20.9 ± 1.5 | 2.8 ± 0.7 |
| CellobioseC5S-Au-SC5DO3A-Gd | 2.0 ± 0.1 | 2.1 ± 0.2 | 16.6 ± 0.9 | 30.4 ± 1.9 | 0.2 ± 0.2 |
| MaltoseC5S-Au-SC11DO3A-Gd | 1.8 ± 0.2 | 3.1 ± 0.2 | 12.8 ± 0.8 | 21.2 ± 1.3 | 3.5 ± 0.9 |
| MaltoseC5S-Au-SC5DO3A-Gd | 1.8 ± 0.2 | 3.6 ± 0.2 | 14.7 ± 0.6 | 25.5 ± 1.6 | 0.8 ± 0.4 |
| Dotarem® | — | — | 3.0 ± 0.2 | — | 3.6 ± 0.8 |
3.2 Monitoring cellular binding of Gd-GNPs at 11.7 T
Before testing the Gd-GNPs on cellular lines, their cytotoxicity was evaluated using the MTT method (ESI, Fig. S5†).57 The cellular viability was more than 80% after incubation for 24 h at concentrations up to 100 μg mL−1 of Gd-GNPs. For the cellular experiments, we selected the Gd-GNPs bearing the SC11DO3A-Gd complex and 5-mercaptopentyl glycosides of β-glucose (Glc-GNP), β-galactose (Gal-GNP), α-mannose (Man-GNP), β-lactose (Lac-GNP), and β-cellobiose (cellobiose-GNP) (Table 1). The selection was firstly based on the excellent behaviour of the GlcC5S-Au-SC11DO3A-Gd GNP (Glc-GNP) as T1-probe in an ex vivo screening (C. Arus et al., personal communication) and in vivo experiments with mouse glioma.48 Secondly, GlcC5S-Au-SC11DO3A-Gd (Glc-GNP, 7.4 ± 0.7 mM−1 s−1) gave a higher ΔT1 (70%) than GlcC5S-Au-SC5DO3A-Gd (60%) in the in vitro labelling of GL261 cells at 11.7 T, in spite of the higher relaxivity value (r1 16.9 mM−1 s−1, Table 1) of the latter Gd-GNP (data not shown).The detection of individual cells in their natural or pathological context by non-invasive techniques is of enormous significance to identify a specific cell population (stem, cancer cells), to follow the progression of diseases, and to evaluate the efficacy of cell therapies in preclinical and clinical studies. Labelling cells with paramagnetic contrast agents presents many drawbacks, but is highly promising and can complement the use of superparamagnetic iron oxide-based nanoparticles when the image darkening (“negative” contrast) is not suitable to study a biological process.5 Two scenarios can be envisaged for labelling cells: to target cell surface receptors and/or to internalize the paramagnetic agent into the cells. Fixed cells were used to study the carbohydrate-mediated targeting of surface receptors. When a cell is fixed, any ongoing biochemical reaction stops, but an increase in their mechanical strength or stability is achieved. This increase helps to preserve the cell morphology, allowing the study of surface receptors. Live cells were also studied with Gd-GNPs. Labelling of live cells with Gd-contrast agents is still a challenge because internalization of Gd-complexes frequently results in quenching of the “positive” contrast.58,59
In order to check whether Gd-GNPs could work as T1 reporters in both scenarios, we have explored the in vitro labelling of different cell lines that express specific receptors at their surface. The selected cells are Burkitt lymphoma cells (Raji and Raji-DC-SIGN), hepatocytes (HepG2), and murine glioma cells (GL261). Raji DC-SIGN (Raji+) cells overexpress the receptor DC-SIGN (Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin), a calcium-dependent and tetrameric lectin which specifically recognizes oligomannosides and Lewis blood group antigens through multivalent protein–carbohydrate interactions.60 Raji negative cells do not express DC-SIGN. However, both Raji and Raji+ cells express a mannose-binding receptor61 that recognizes mannose, glucose, and glucosamine. Monosaccharides with equatorial 4-hydroxyl groups (e.g. mannose and glucose) preferentially bind to mannose receptors with respect to galactose that has an axial hydroxyl group in position 4 of the pyranose ring.62 In a previous study, we have shown that fluorescent GNPs coated with multiple copies of oligomannosides are taken up and internalized preferentially by Raji+ with respect to Raji cells.63
HepG2 cells are known to express the β-galactose/N-acetylgalactosamine binding lectin named asialoglycoprotein receptor.64 This hetero-oligomeric receptor is expressed almost exclusively in hepatic parenchymal cells and binds specifically to galactosyl-terminal glycoproteins. The role of multivalence in the binding of synthetic oligosaccharides to this lectin has been demonstrated by the pioneer work of Lee et al.65 The multimerization of β-galactoside moieties on gold nanoparticles should thus ensure a good interaction with HepG2 cells.
The murine glioma cells (GL261) were used to generate the brain tumour in mice for the in vivo evaluation of the Gd-based glyconanoparticles.48 These cells were selected to compare both the in vivo and in vitro performance. In addition, the heterogeneity of glucose metabolism of glioma cells may confer on the Glc-GNP a prognostic significance.66
Raji, Raji+, and GL261 lines are supposed to overexpress GLUT transporters67 typical of tumour-derived cells. The Gd-GNPs incorporating mannose (Man-GNP) and galactose (Gal-GNP) are expected to target preferentially Raji+ and HepG2, respectively, while glucose (Glc-GNP) should label all cell lines at a similar level.
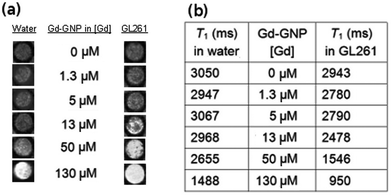 | ||
| Fig. 2 (a) T1-weighted images of an aqueous solution of Glc-GNP (left) and the incubated fixed GL261 cells (right) at different Gd concentrations; and (b) the corresponding T1 values of Glc-GNP in water and incubated with GL261 cells at 11.7 T. | ||
To evaluate the selectivity of the Gd-GNPs, 105–106 Raji, Raji+, HepG2 or GL261 fixed cells were incubated for 1 hour at 37 °C in the presence of paramagnetic Glc-, Gal-, and Man-GNPs at 130 μM (19.7 μg mL−1) concentration of Gd. All paramagnetic GNPs at this concentration significantly decreased the T1 of cell images. When the cells were fixed and incubated with 130 μM Dotarem®, T1-weighted images of the cell pellet did not reveal contrast enhancement. From the T1-weighted images of the cellular phantoms (Fig. 3a), it can be qualitatively assessed that HepG2 and GL261 bind preferably Gal-GNP and Glc-GNP, respectively, while Raji/Raji+ cells bind Glc- and Gal-GNP indistinctly. A more exact picture is obtained by quantification of the percentage change of the T1 (%ΔT1 = (T1cells − T1cells+Gd-GNPs) × 100/T1cells) obtained from the T1 maps of control cell pellets and those of Gd-GNPs incubated cells (Fig. 3b). These data indicate that Glc-GNP induces a similar ΔT1 (70%) in Raji, Raji+, and GL261 cells, while the reduction of T1 in HepG2 cells is only 37%. This selectivity is clearly observed at 50 μM (7.6 μg mL−1) concentration of Gd with a T1 reduction of 2% for Glc-GNP (data not shown) and 29% for Gal-GNP (Table S3†). As expected,68 HepG2 cells seem to take up preferably Gal-GNP with a ΔT1 of 66% followed by Man-GNP with 54%. Man-GNP binds preferentially Raji and Raji+ cells with respect to the other cells (Fig. 3b). Although the T1-weighted images of Raji and Raji+ qualitatively show a clear preference of Raji+ for Man-GNPs (Fig. 3a), statistical analysis of the data (n = 4) indicates that this difference is not significant (p > 0.05). This can be explained by the binding of the Man-GNP mainly to mannose receptors instead of the DC-SIGN receptor.
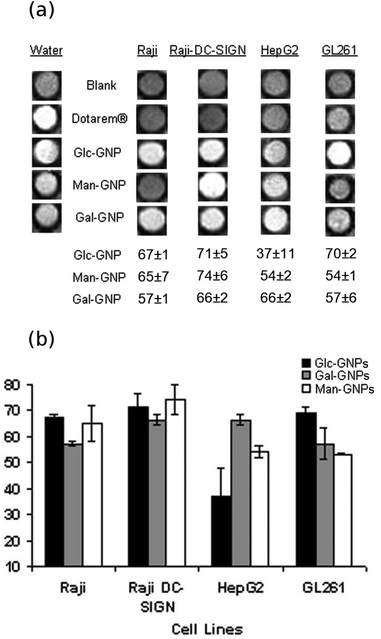 | ||
| Fig. 3 (a) T1-weighted images of fixed cells at 11.7 T and the percentage change of T1 (%ΔT1) after incubation with Dotarem® or Gd-GNP at 130 μM (17.9 μg mL−1) concentration of Gd and (b) bar diagrams of the percentage change of T1 (%ΔT1) of the cells after incubation with Gd-GNPs. Each value and error bar in the plot represent the mean and standard deviation from independent experiments (n = 2–5). | ||
To test the specificity of Glc-GNP for glucose-binding proteins (receptors/transporters) a competition experiment was performed. Pre-incubation of fixed GL261 cells with D-glucose (1 M) followed by treatment with Gd-GNP showed T1 values slightly higher (1826 ± 37 ms, ΔT1 28%) than those of cells incubated only with Glc-GNP (1379 ± 69 ms, ΔT1 45%), pointing out that a high concentration of glucose moderately blocks the receptor. In contrast, pre-incubation with gold nanoparticles 100%-coated with 3 mM concentration of glucose (GlcC5S-Au, Scheme 1) showed similar T1 values (2622 ± 43 ms) to the control cells (2537 ± 29 ms). This result can be attributed to the multivalent presentation of glucose on GNP associated with an increase of local concentration, which enhances the avidity of glucose for the receptor.
In summary, Gd-GNPs bind cell receptors, reducing significantly the longitudinal relaxation times. The presence of sugars on the paramagnetic GNPs modulates the labelling of selected cell lines with ΔT1 between 37 and 74%. A clear selectivity of HepG2 for Gal-GNP and Man-GNP versus Glc-GNP was observed, while Raji/Raji+ cells preferentially bind Man-GNP in comparison with the other cells. Taking into account that tumour derived cell lines overexpress sugar receptors to maintain their fast-growing metabolism, the difference in binding observed here can be considered significant. The results demonstrate the potential of the sugar/Gd-GNPs as a tool for mapping the carbohydrate-binding features of selected cells and disclosing the sugar-binding receptors at the cell surface.
We have also tested Gd-GNPs coated with the disaccharide lactose (galactopyranosylβ1-4glucose) that is recognized by the asialoglycoprotein,64 and cellobiose (glucopyranosylβ1-4glucose), a non-naturally occurring disaccharide in mammalian cells. Lac-GNP incorporating the disaccharide lactose reduced the T1 values by 58% in HepG2 cells and by 52% in GL261 cells, while the cellobiose-GNP produced a 66% decrease of T1 in HepG2 (data not shown). The origin of the decrease observed for cellobiose in HepG2 cells is probably due to the terminal glucose, but is unclear. It would be worth a deeper investigation of Gd-GNPs coated with more complex sugars.
In a general overview, the incubation of cells alive with selected Gd-GNP at 50 μM (7.6 μg mL−1) concentration of Gd gave a range of %ΔT1 values between 8 and 30%, smaller than those observed for the fixed cells (24–60%) (Table S3†). For example, incubation of live HepG2 cells with Gal-GNP at 50 μM concentration of Gd reduced the T1 values by 20%, which is slightly lower than the 30% for fixed cells. However, live GL261 incubated with Glc-GNP gave ΔT1 (∼27%) similar to fixed cells (Table S3†). Only a significant difference in the %ΔT1 values between live and fixed was observed in Raji/Raji+ cells incubated with Man-GNP at 50 μM (Fig. 4a). The lower contrast reduction observed for live cells in comparison with the fixed cells at the same incubation concentration may be due to different reasons. One is that live cells can excrete part of the nanoparticles, unlike fixed cells. Furthermore, in fixed cells high local “concentrations” at the cell surface can increase the contrast, while in live cells GNPs are not confined in the external membrane, but they may be distributed in different cellular compartments. In addition, phenomena of quenching inside the cells can reduce the contrast as already mentioned.58,59
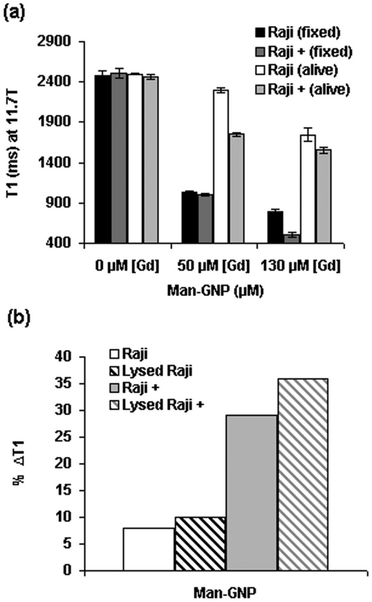 | ||
| Fig. 4 (a) T1 values of fixed and live Raji and Raji+ cells incubated with Man-GNP at 50 μM and 130 μM concentrations of Gd; and (b) comparison of %ΔT1 of live and lysed Raji and Raji+ cells after incubation with Man-GNP at 50 μM concentration of Gd. A representative value of several experiments is shown. Error bars correspond to the curve-fitting of T1 map image intensities. | ||
It is worth noting that the difference in ΔT1 between Raji and Raji+ cells for Man-GNP is higher in live (8% vs. 29%) than in fixed cells (58% vs. 60%). In any case, the observed decrease of T1 values in live cells is in the range of those that have been reported in the literature for the DNA/Gd loaded gold nanoparticles.29 The significant difference observed between fixed and live Raji/Raji+ cells may depend on intracellular mechanisms in live cells. To investigate the possibility of Gd-GNPs intracellular entrapment and quenching,58 the same live Raji/Raji+ pellets previously incubated with Man-GNP were lysed in HEPES buffer and the T1 values were measured at 11.7 T. The ΔT1 of both live and lysed Raji cells yielded the same values (8/10%, Fig. 4b) as is also the case of HepG2 cells incubated with Gal-GNP (Table S3†). In the case of live/lysed Raji+, the difference is slightly higher (29/36%). These results suggest that no significant intracellular quenching in live cells seems to take place.
Many factors seem to influence the interaction of cells with glyconanoparticles (cell type and conditions, Gd-GNPs concentrations, and sugar type) that deserved further investigations. However, in this study we have demonstrated that simple sugars are excellent molecules to modulate the uptake of nanoparticles by cells. These results confirm that the novel sugar/Gd coated gold nanoparticles are efficient T1-probes for labelling tumour-derived cell lines. The sugar molecules confer biocompatibility, stability and selectivity as well as cell permeability on the paramagnetic nanoparticles. The application of these new probes can be extended to all types of cells because glucose, galactose, and mannose are efficient sources for the catabolism of all types of cells, not only tumour cells.73 Furthermore, the targeting properties of the sugar/Gd-coated nanoparticles could be improved by incorporating into the gold nanocluster complex oligosaccharides that are specific ligands of cell surface receptors.63 A similar study by Huang and co-workers on sugar-functionalized iron oxide nanoparticles (T2-probes) has been recently reported,41 but negative contrast agents have limitations for in vivo studies, and the development of biocompatible and permeable positive contrast agents is today an urgent need for the in vivo applications.
4. Conclusions
We have prepared sugar/Gd-coated gold nanoparticles and demonstrated their efficiency as permeable and highly biocompatible cellular reporter probes for MRI at high magnetic fields. Carbohydrate-coated paramagnetic gold nanoparticles take advantage of the affinity of cells for simple sugars. Moreover, the polyvalent presentation of the sugars on the gold surface increases their affinity for lectins and sugar transporters and therefore the concentration of Gd cations at the cell surface. With an average number of 20 Gd atoms per nanoparticle, relaxivity values ranging from 7 to 18 mM−1 s−1 have been obtained. A large reduction in T1 upon incubation of cells with sugar-GNPs has been achieved. We have also demonstrated that by changing the sugar on the nanoparticles surface significant differences in cellular uptake are obtained. These results stress the benefits of using sugars41,63,70–72 as an alternative to oligonucleotides,29,74–76 peptides, or polyethylene glycol molecules to produce nanoparticles for cellular labelling. The evaluation of Gd-GNPs in an ex vivo post-mortem MRI study and further in vivo experiments confirm Gd-GNPs as MRI agents for the detection of glioma in mice.77Acknowledgements
This study was supported by the Spanish Ministry of Science and Innovation (MICINN, grants CTQ2008-04638 and CTQ2011-27268), the Department of Industry of the Basque Country (grant ETORTEK 2009 and 2011) and the Department of Education, Universities and Research of the Basque Government (Project PI2012-46). A. I. thanks the Basque Government for a predoctoral grant. We thank Irati Markuerkiaga and Sandra Plaza for technical assistance in MRI experiments.Notes and references
- A. C. Elsaesser and V. Howard, Adv. Drug Delivery Rev., 2012, 64, 129 CrossRef CAS.
- M. A. Dobrovolskaia and S. E. McNeil, Nat. Nanotechnol., 2007, 2, 469 CrossRef CAS.
- S. Rana, A. Bajaj, R. Mout and V. M. Rotello, Adv. Drug Delivery Rev., 2012, 64, 200 CrossRef CAS.
- H. S. Choi, W. Liu, F. Liu, K. Nasr, P. Misra, M. G. Bawendi and J. V. Frangioni, Nat. Nanotechnol., 2010, 5, 42 CrossRef CAS.
- N. Muja and J. W. M. Bulte, Prog. Nucl. Magn. Reson. Spectrosc., 2009, 55, 61 CrossRef CAS.
- E. Terreno, D. Delli Castelli, A. Viale and S. Aime, Chem. Rev., 2010, 110, 3019 CrossRef CAS.
- S. Aime, Z. Baranyai, E. Gianolio and E. Terreno, in Molecular and Cellular MR Imaging, ed. M. M. J. Modo and J. W. M. Bulte, Taylor and Francis Group (CRC Press), Boca Raton, FL, 2007, p. 37 Search PubMed.
- R. C. Strauch, D. J. Mastarone, P. A. Sukerkar, Y. Song, J. J. Ipsaro and T. J. Meade, J. Am. Chem. Soc., 2011, 133, 16346 CrossRef CAS.
- J. L. Major and T. J. Meade, Acc. Chem. Res., 2009, 42, 893 CrossRef CAS.
- E. Rodriguez, M. Nilges, R. Weissleder and J. W. Chen, J. Am. Chem. Soc., 2010, 132, 168 CrossRef CAS.
- P. Caravan, Acc. Chem. Res., 2009, 42, 851 CrossRef CAS.
- T. Yamane, K. Hanaoka, K. Muramatsu, Y. Adachi, Y. Miyashita, Y. Hirata and T. Nagano, Bioconjugate Chem., 2011, 22, 2227 CrossRef CAS.
- I. S. Nolte, S. Gungor, R. Erber, E. Plaxina, J. Scharf, B. Misselwitz, L. Gerigk, H. Przybilla, C. Groden and M. A. Brockmann, Magn. Reson. Med., 2008, 59, 1014 CrossRef CAS.
- T. D. Henning, O. Saborowski, D. Golovko, S. Boddington, J. S. Bauer, Y. Fu, R. Meier, H. Pietsch, B. Sennino, D. M. McDonald and H. E. Daldrup-Link, Eur. Radiol., 2007, 17, 1226 CrossRef.
- E. Vucic, H. M. H. F. Sanders, F. Arena, E. Terreno, S. Aime, K. Nicolay, E. Leupold, M. Dathe, N. A. J. M. Sommerdijk, Z. A. Fayad and W. J. M. Mulder, J. Am. Chem. Soc., 2009, 131, 406 CrossRef CAS.
- D. A. Sipkins, D. A. Cheresh, M. R. Kazemi, L. M. Nevin, M. D. Bednarski and K. C. P. Li, Nat. Med., 1998, 4, 623 CrossRef CAS.
- C. H. Reynolds, N. Annan, K. Beshah, J. H. Huber, S. H. Shaber, R. E. Lenkinski and J. A. Wortman, J. Am. Chem. Soc., 2000, 122, 8940 CrossRef CAS.
- A. J. L. Villaraza, A. Bumb and M. W. Brechbiel, Chem. Rev., 2010, 110, 2921 CrossRef CAS.
- Z. Cheng, D. L. J. Thorek and A. Tsourkas, Angew. Chem., Int. Ed., 2010, 49, 346 CrossRef CAS.
- E. Wiener, M. W. Brechbiel, H. Brothers, R. L. Magin, O. A. Gansow, D. A. Tomalia and P. C. Lauterbur, Magn. Reson. Med., 1994, 31, 1 CrossRef CAS.
- H. B. Na, I. C. Song and T. Hyeon, Adv. Mater., 2009, 21, 2133 CrossRef CAS.
- J.-K. Hsiao, C. P. Tsai, T. H. Chung, Y. Hung, M. Yao, H. M. Liu, C. Y. Mou, C. S. Yang, Y. C. Chen and D. M. Huang, Small, 2008, 4, 1445 CrossRef CAS.
- P.-J. Debouttière, S. Roux, F. Vocanson, C. Billotey, O. Beuf, A. Favre-Réguillon, Y. Lin, S. Pellet-Rostaing, R. Lamartine, P. Perriat and O. Tillement, Adv. Funct. Mater., 2006, 16, 2330 CrossRef.
- J.-L. Bridot, A.-C. Faure, S. Laurent, C. Rivière, C. Billotey, B. Hiba, M. Janier, V. Josserand, J.-L. Coll, L. V. Elst, R. Muller, S. Roux, P. Perriat and O. Tillement, J. Am. Chem. Soc., 2007, 129, 5076 CrossRef CAS.
- L. Moriggi, C. Cannizaro, E. Dumas, C. R. Mayer, A. Ulianov and L. Helm, J. Am. Chem. Soc., 2009, 131, 10828 CrossRef CAS.
- L. Frullano and T. J. Meade, J. Biol. Inorg. Chem., 2007, 12, 939 CrossRef CAS.
- E. Garanger, E. Aikawa, F. Reynolds, R. Weissleder and L. Josephson, Chem. Commun., 2008, 4792 RSC.
- Y. T. Lim, M. Y. Cho, B. S. Choi, J. M. Lee and B. H. Chung, Chem. Commun., 2008, 40, 4930 RSC.
- Y. Song, X. Y. Xu, K. W. MacRenaris, X.-Q. Zhang, C. A. Mirkin and T. J. Meade, Angew. Chem., Int. Ed., 2009, 48, 9143 CrossRef CAS.
- M. F. Warsi, R. W. Adams, S. B. Duckett and V. Chechik, Chem. Commun., 2010, 46, 451 RSC.
- J. A. Park, H. K. Kim, J. H. S. Kim, W. Jeong, J. C. Jung, G. H. Lee, J. Lee, Y. Chang and T. J. Kim, Bioorg. Med. Chem. Lett., 2010, 20, 2287 CrossRef CAS.
- D. Artemov, J. Cell. Biochem., 2003, 90, 518 CrossRef CAS.
- H. Yang, Y. Zhuang, Y. Sun, A. Dai, X. Shi, D. Wu, F. Li, H. Hu and S. Yang, Biomaterials, 2011, 32, 4584 CrossRef CAS.
- A. Varki and N. Sharon, in Essentials in Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart and M. E. Etzler, Cold Spring Harbor, New York, 2nd edn, 2009, p. 1 Search PubMed.
- T. Feizi and B. Mulloy, Curr. Opin. Struct. Biol., 2003, 13, 602 CrossRef CAS.
- C. R. Bertozzi and L. L. Kiessling, Science, 2001, 291, 2357 CrossRef CAS.
- T. M. Allen, Adv. Drug Delivery Rev., 1994, 13, 285 CrossRef CAS.
- M. Monsigny, A. C. Roche, P. Midoux and R. Mayer, Adv. Drug Delivery Rev., 1994, 14, 1 CrossRef CAS.
- M. Marradi, F. Chiodo, I. García and S. Penadés, Chem. Soc. Rev., 2013 10.1039/c2cs35420a.
- L. Cipolla, M. P. Gregori and W. So, Curr. Med. Chem., 2011, 18, 1002 CrossRef CAS and references therein.
- K. El Boubbou, D. C. Zhu, C. Vasileiou, B. Borhan, D. Prosperi, W. Li and X. Huang, J. Am. Chem. Soc., 2010, 132, 4490 CrossRef CAS.
- H. Lis and N. Sharon, Chem. Rev., 1998, 98, 637 CrossRef CAS.
- Y. C. Lee and R. T. Lee, Acc. Chem. Res., 1995, 28, 321 CrossRef CAS.
- J. M. de la Fuente, A. G. Barrientos, T. C. Rojas, J. Rojo, J. Cañada, A. Fernández and S. Penadés, Angew. Chem., Int. Ed., 2001, 40, 2257 CrossRef.
- M. Marradi, M. Martín-Lomas and S. Penadés, Adv. Carbohydr. Chem. Biochem., 2010, 64, 211 CrossRef CAS.
- J. M. de la Fuente and S. Penadés, Glycoconjugate J., 2004, 21, 149 CrossRef CAS.
- O. Warburg, Science, 1956, 123, 309 CAS.
- M. Marradi, D. Alcántara, J. M. de la Fuente, M. L. García-Martín, S. Cerdán and S. Penadés, Chem. Commun., 2009, 3922 RSC.
- H. Hatanaka, Biochim. Biophys. Acta, 1974, 355, 77 Search PubMed.
- D. Högemann, V. Ntziachristos, L. Josephson and R. Weissleder, Bioconjugate Chem., 2002, 13, 116 CrossRef.
- H. Lee, E. Sun, D. Ham and R. Weissleder, Nat. Med., 2008, 14, 869 CrossRef CAS.
- M. J. Hostetler, J. E. Wingate, C.-J. Zhong, J. E. Harris, R. W. Vachet, M. R. Clark, J. D. Londono, S. J. Green, J. J. Stokes, G. D. Wignall, G. L. Glish, M. D. Porter, N. D. Evans and R. W. Murray, Langmuir, 1998, 14, 17 CrossRef CAS.
- M. J. Hostetler, A. C. Templeton and R. W. Murray, Langmuir, 1999, 15, 3782 CrossRef CAS.
- A. G. Barrientos, J. M. de la Fuente, T. C. Rojas, A. Fernández and S. Penadés, Chem.–Eur. J., 2003, 9, 1909 CrossRef CAS.
- R. Ojeda, J. L. de Paz, A. G. Barrientos, M. Martín-Lomas and S. Penadés, Carbohydr. Res., 2007, 342, 448 CrossRef CAS.
- O. Martínez-Ávila, K. Hijazi, M. Marradi, C. Clavel, C. Campion, C. Kelly and S. Penadés, Chem.–Eur. J., 2009, 15, 9874 CrossRef.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55 CrossRef CAS.
- E. Terreno, S. G. Crich, S. Belfiore, L. Biancone, C. Cabella, G. Esposito, A. D. Manazza and S. Aime, Magn. Reson. Med., 2006, 55, 491 CrossRef CAS.
- E. Gianolio, F. Arena, G. J. Strijkers, K. Nicolay, A. Högset and S. Aime, Magn. Reson. Med., 2011, 65, 212 CrossRef CAS.
- Y. Guo, H. Feinberg, E. Conroy, D. A. Mitchell, R. Alvarez, O. Blixt, M. E. Taylor, W. I. Weis and K. Drickamer, Nat. Struct. Mol. Biol., 2004, 11, 591 CAS.
- M. Kato, K. J. McDonald, S. Khan, I. L. Ross, S. Vuckovic, K. Chen, D. Munster, K. P. A. MacDonald and D. N. Hart, Int. Immunol., 2006, 18, 857 CrossRef CAS.
- S. V. Su, P. Hong, S. Baik, O. A. Negrete, K. B. Gurney and B. Lee, J. Biol. Chem., 2004, 279, 19122 CrossRef CAS.
- B. Arnáiz, O. Martínez-ávila, J. M. Falcon-Perez and S. Penadés, Bioconjugate Chem., 2012, 23, 814 CrossRef.
- J. Lunney and G. Ashwell, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 341 CrossRef CAS.
- Y. C. Lee, R. R. Townsend, M. R. Hardy, J. Lönngren, J. Arnarp, M. Haraldsson and H. Lönn, J. Biol. Chem., 1983, 258, 199 CAS.
- C. E. Griguer, C. R. Oliva and G. Y. Gillespie, J. Neuro-Oncol., 2005, 74, 123 CrossRef CAS.
- M. Mueckler, Eur. J. Biochem., 1994, 219, 713 CrossRef CAS.
- K. Kawaguchi, M. Kuhlenschmidt, S. Roseman and Y. C. Lee, J. Biol. Chem., 1981, 256, 2230 CAS.
- S. D. Conner and S. L. Schmid, Nature, 2003, 422, 37 CrossRef CAS.
- J. M. de la Fuente, D. Alcántara and S. Penadés, IEEE Trans. Nanobiosci., 2007, 6, 275 CrossRef.
- M. Moros, B. Hernáez, E. Garet, J. T. Dias, B. Sáez, V. Grazu, A. Gónzalez-Férnandez, C. Alonso and J. M. de la Fuente, ACS Nano, 2012, 6, 1565 CrossRef CAS.
- J. Gallo, N. Genicio and S. Penadés, Adv. Healthcare Mater., 2012, 1, 302 CrossRef CAS.
- O. Warburg, F. Wind and E. Negelein, J. Gen. Physiol., 1927, 8, 519 CrossRef CAS.
- M. D. Massich, D. A. Giljohann, A. L. Schmucker, P. C. Patel and C. A. Mirkin, ACS Nano, 2010, 4, 5641 CrossRef CAS.
- N. L. Rosi, D. A. Giljohann, C. S. Thaxton, A. K. R. Lytton-Jean, M. S. Han and C. A. Mirkin, Science, 2006, 312, 1027 CrossRef CAS.
- S. Bamrungsap, T. Chen, M. I. Shukoor, Z. Chen, K. Sefah and Y. Chen, ACS Nano, 2012, 6, 3974 CrossRef CAS.
- A. P. Candiota, M. Acosta, R. Simoes, T. Delgado-Goni, S. Lope-Piedrafita, A. Irure, M. Marradi, O. Bomati-Miguel, N. Miguel-Sancho, I. Abasolo, S. Schwartz Jr., J. Santamaria, S. Penades and C. Arus, submitted.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of neoglycoconjugates, preparation of 100% sugar-coated glyconanoparticles, chemical properties of Gd-based paramagnetic glyconanoparticles (Gd-GNPs), determination of relaxivity values, 17O NMR experiments, Dynamic Light Scattering of selected nanoparticles, cytotoxicity assays, MRI phantoms. See DOI: 10.1039/c3bm60032g |
| This journal is © The Royal Society of Chemistry 2013 |