Synthesis, characterization, and in vitro evaluation of two synergistic anticancer drug-containing hepatoma-targeting micelles formed from amphiphilic random copolymer†
Jianfeng
Wang
,
Cui
Yin
,
Guping
Tang
,
Xianfu
Lin
and
Qi
Wu
*
Department of Chemistry, Zhejiang University, Hangzhou, 310027, People's Republic of China. E-mail: llc123@zju.edu.cn; Fax: +86 571 8795 2618; Tel: +86 571 8795 1588
First published on 1st May 2013
Abstract
In this study we fabricated and characterized novel galactose-functionalized multidrug-containing nanoparticles, and in vitro evaluated their enhanced anti-tumorous cell cytotoxicity and hepatoma-targeting ability for the controlled delivery of two synergistic anticancer drugs. A chemo-enzymatic synthesis strategy was used to prepare a galactose-functionalized amphiphilic random copolymer containing cytarabine (Ara-C) and fluorodeoxyuridine (FUDR). The formed nanoparticles were characterized for their critical aggregation concentration (CAC), morphology, cellular uptake, cell cytotoxicity, hepatoma targeting ability and controlled drug release. The micellization ability of the galactose-functionalized amphiphilic random copolymer was confirmed. In vitro drug release studies showed that the two anticancer agents could be simultaneously released from the nanoparticles. Cellular uptake assay and cytotoxicity tests demonstrated that these nanoparticles could be effectively internalized by HepG2 cells and had an evident targeting function through the selective recognition of galactose pendants of the copolymer by ASGP-R of HepG2 cells. Furthermore, the enhanced anti-tumorous cell cytotoxicity indicated the combination of Ara-C, FUDR and D-galactose in one copolymer may have a better synergistic effect. This encouraging finding illustrated that the D-galactose functionalized nanoparticles could be used as a novel potential hepatoma-targeting multidrug delivery vehicle.
1. Introduction
During the past decade, a great deal of research has concentrated on new cancer therapeutics, among which, polymer therapeutics, including rationally designed macromolecular drugs, polymer–drug and polymer–protein conjugates, and polymeric micelles containing covalently bound drugs are the most attractive.1,2 Polymer delivery systems including polymer–drug conjugates and polymeric micelles have proven to be useful. A few combinations of a limited number of chemotherapeutic drugs and polymer delivery systems have been tested in clinical and preclinical trials.3–8Polymer conjugation could radically change the pharmacokinetics of the bound drug, prolong drug circulation in the blood and target tumors passively by an enhanced permeability and retention (EPR) effect.9,10 Polymeric drug carriers can also be modified with moieties such as RGD,11 antibody,12 glycyrrhizin,13 folate,14 and transforming growth factor (TGF)-B15 to directly target tumour cells. Among the receptors of these ligands, asialoglycoprotein receptor (ASGP-R) is the most promising for drug targeting due to its abundant expression on the surface of the hepatocytes and the high affinity with galactose as a ligand.16 For this reason, incorporations of carbohydrates into drug delivery systems have attracted considerable interest,16–19 and many articles have reported galactose-based copolymers or nanoparticles for targeting hepatoma cells.20–22
As most anticancer drugs are water-insoluble, highly toxic and have some resistance mechanisms, limiting their clinical applications, drug combination therapy especially multi-agent combinational therapy has attracted much more attention in recent years.23–26 Duncan et al. synthesized a HPMA (i.e. N-(2-hydroxypropyl)methacrylamide)-based polymer–drug conjugate carrying both doxorubicin (Dox) and aminoglutethimide (AGM) and the synergistic cytotoxic effects of AGM and Dox in conjugated form effectively enhanced the efficiency compared to a HPMA–Dox single-agent copolymer.27,28 In our study, we chose Cytarabine (Ara-C)29 and fluorodeoxyuridine (FUDR)30,31 as the model drugs to investigate drug combination therapy. As two kinds of important clinical anticancer drugs, it has been verified that the combination of Ara-C and FUDR showed synergistic effects against a series of cell lines including leukemia, ovarian and breast cancer cells.32–34 We have incorporated Ara-C and FUDR into polymeric nanoparticles and investigated their in vitro drug delivery characters in our previous work.35 However, there were several existing drawbacks such as the unsatisfied solubility, the unknown synergistic activity of the duplex drugs and the lack of desirable targeting ability, which need to be considered in further studies.
Herein, we designed a novel nanocarrier self-assembled from an amphiphilic random copolymer with Ara-C and FUDR as the bound model drugs and galactose as the hepatoma targeting ligand. The galactose-functionalized random copolymers were synthesized via a chemo-enzymatic strategy and IR, NMR, and gel permeation chromatography (GPC) were used to determine their well-defined structures. The micellization ability and properties such as critical aggregation concentration (CAC), morphology and size distribution were characterized and measured by fluorescence probe technology, transmission electron microscopy (TEM) and dynamic light scattering (DLS). The in vitro drug release behavior was studied in a buffer modeling extracellular environment. Most importantly, we aimed to investigate the hepatoma-targeting function of the micellar nanoparticles and whether “drug combination therapy” could improve the efficacy against HepG2 human hepatoma cells by a MTT assay.
2. Results and discussion
2.1. Synthesis of amphiphilic random copolymer
In this study, a galactose-functionalized amphiphilic random copolymer poly(VAF-co-VSC-co-VGA) containing cytarabine and fluorodeoxyuridine was easily prepared by combining enzymatic transesterifications and radical polymerization. This two-step chemo-enzymatic synthetic route was shown in Scheme 1. According to the literature,18–20 enzymes play an important role in the selective structural modification of drugs and sugars. By enzymatic selective acylation of Ara-C, FUDR and D-galactose with divinyl dicarboxylates, polymerizable vinyl derivatives (VSC, VAF, and VGA) were facilely synthesized. The resulting vinyl derivatives could be subsequently used as comonomers for further synthesis of the copolymer with drugs and sugar as pendants.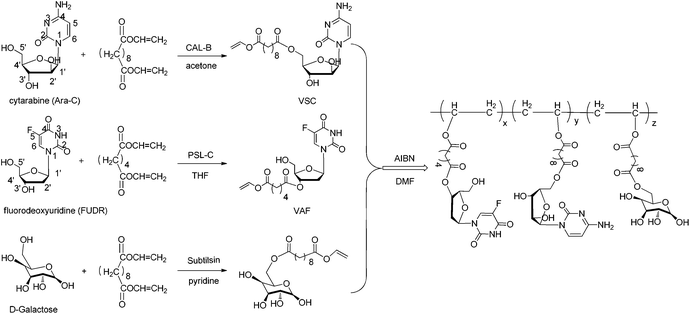 | ||
| Scheme 1 Chemo-enzymatic synthesis of a galactose-functionalized multidrug copolymer containing cytarabine and fluorodeoxyuridine. | ||
The galactose-functionalized multidrug-containing amphiphilic random copolymer poly(VAF-co-VSC-co-VGA) was prepared by radical copolymerization of vinyl drug derivatives with a galactose derivative at 70 °C in DMF using AIBN as the initiator. We chose different divinyl dicarboxylates as the acylating donors for FUDR, Ara-C and D-galactose under optimized reaction conditions according to the literature.36–38 The resulting copolymer was characterized by FTIR, 1H NMR and GPC. The absence of vinyl proton signals (1H NMR: δ 7.22, 4.88, 4.65) and the existence of FUDR (1H NMR: δ 11.98, 8.07, 6.15), Ara-C (1H NMR: δ 7.56, 7.27, 7.12) and galactose (1H NMR: δ 6.66, 6.31, 5.30–2.85) moieties in the 1H NMR spectrum confirmed the success of the copolymerization (Fig. S2†). Also, the FTIR spectrum (Fig. S1†) of the copolymer showed that vinyl group absorption present in the comonomers disappeared in the corresponding copolymer. The molar percentage of Ara-C and FUDR was respectively about 39% and 35% in the copolymer by calculating the ratio of the integral area of peaks corresponding to cytarabine, fluorodeoxyuridine and galactose in the 1H NMR spectrum of the copolymer. The number-average molecular weight (Mn) of poly(VAF-co-VSC-co-VGA) measured by GPC was about 4.34 × 104, and the polydispersity (Mw/Mn) was 1.41. Moreover, the galactose-functionalized single-drug-containing amphiphilic random copolymers poly(VAF-co-VGA) and poly(VSC-co-VGA) were similarly prepared and fully characterized by the same methods. The molar percentage of Ara-C in poly (VSC-co-VGA) was 70%, while the molar percentage of FUDR in poly(VAF-co-VGA) was 55%.
2.2. Nanoparticle preparation and structure characterization
Using a dialysis method, micellar nanoparticles were prepared from the resulting amphiphilic random copolymer. As the copolymer contained hydrophobic main chains and hydrophilic pendants (drugs and galactose), water was used as the polar solvent to trigger the self-assembly of the copolymer. We chose DMSO as the cosolvent as it could not only dissolve hydrophobic and hydrophilic segments of the copolymer but was also miscible with water. A certain amount of the copolymer was dissolved in DMSO, and then the solution was mixed with water so the DMSO would diffuse into water to induce microphase separation of the hydrophobic and hydrophilic segments. The hydrophobic parts were tucked in the interior of an assembly and the hydrophilic parts were exposed to the bulk solvent, which are shown in Fig. 1. As one polymer molecule of poly(VAF-co-VSC-co-VGA) with such little Mn (4.34 × 104) was impossible to constitute such a big micelle, it is more reasonable to consider it as “multi-molecule micelles” or “large compound micelles”.39,40 Actually, the backbone of the copolymer is flexible, which can easily accommodate the conformational change during phase segregation. Thus, both intra- and intermolecular microphase segregation may occur rapidly, inducing aggregation of the hydrophobic backbone of several copolymer molecules to form a compact core. Meanwhile the hydrophilic groups at the surface will be increased to stabilize the whole multimolecular micelles.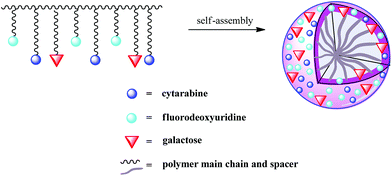 | ||
| Fig. 1 Schematic representation of the self-assembly of a galactose-functionalized multidrug copolymer. | ||
To confirm the self-assembly of the copolymer poly(VAF-co-VSC-co-VGA), the critical aggregation concentration (CAC) value was determined by fluorescence spectroscopy using pyrene as the hydrophobic fluorescent probe. The fluorescence emission spectra of pyrene against various poly(VAF-co-VSC-co-VGA) concentrations at a fixed excitation wavelength of 339 nm are shown in Fig. 2a. The fluorescence intensity increased with increasing copolymer concentration, due to the fact that pyrene has a strong hydrophobic character with a very small absorption in water, and the absorption increases when pyrene is transferred into a hydrophobic environment. This indicated that the amphiphilic random copolymer does indeed form core–shell aggregates in water and pyrene was preferentially partitioned into the less polar microdomains of the aggregates. From the emission spectra of pyrene, the intensity ratio of the third band (I384) to the first band (I373) was calculated. The ratio value was very sensitive to the polarity of the medium surrounding the pyrene molecule.41 From the intensity ratio against the logarithm of poly(VAF-co-VSC-co-VGA) concentrations (Fig. 2b), it could be observed that I384/I373 was almost unchanged at low concentrations, while an abrupt increase occurred at a certain concentration, which indicated the formation of aggregates and the transfer of pyrene into the hydrophobic interior of the aggregates. The CAC value of the copolymer poly(VAF-co-VSC-co-VGA) in water was about 1 mg L−1. The low CAC value allowed their use in very dilute aqueous media such as body fluids.
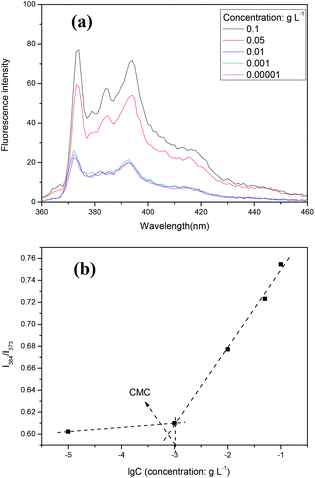 | ||
| Fig. 2 (a) Fluorescence emission spectra of pyrene/nanoparticles against concentration of nanoparticles. (b) The fluorescence intensity ratio (I384/I373) in the emission spectra (a) against the logarithm of the concentration of nanoparticles. | ||
The morphology of the nanoparticles self-assembled from the amphiphilic random copolymer was investigated by transmission electron microscopy (TEM). The TEM image showed that the micelles were well dispersed as individual round spheres in the aqueous phase. The average diameter of the nanoparticles was about 500 nm by visual (Fig. 3a). The size distribution of the nanoparticles was also measured by dynamic light scattering (DLS) analysis. The diameter of the nanoparticles was 472 ± 45 nm in the aqueous solution with a narrow distribution (Fig. 3b). The size of the micelles measured by DLS was in agreement with the TEM result. Compared with the nanoparticles formed from the copolymer poly(VSC-co-VAF) without the pendant of D-galactose,35 we can find the incorporation of D-galactose has an interesting and positive effect on the morphology and size distribution of the formed nanoparticles. The spherical nanoparticles of the copolymer poly(VAF-co-VSC-co-VGA) with the D-galactose branch were dispersed much better, and also had a larger size (472 ± 45 nm vs. 133 ± 28 nm), as well as a much narrower distribution than nanoparticles from the copolymer of poly(VAF-co-VSC) (PDI = 0.218 vs. PDI = 0.529). This phenomenon could be explained in that the incorporation of D-galactose into the random terpolymers significantly improved the water solubility of the micelles as carbohydrate is highly hydrophilic and exposed to the water environment. Thus the micelles were dispersed well in water with a narrower size distribution but a larger size. In addition, the larger molecular weight of poly(VAF-co-VSC-co-VGA) (Mn = 4.34 × 104) also made a contribution to a larger size of nanoparticles from poly(VAF-co-VSC-co-VGA) than nanoparticles from the copolymer of poly(VAF-co-VSC) (Mn = 5012).
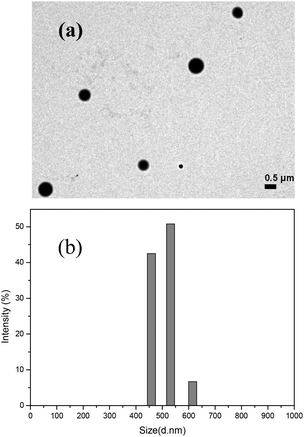 | ||
| Fig. 3 (a) TEM image of nanoparticles formed from poly(VAF-co-VSC-co-VGA). (b) Size characterization of nanoparticles formed from poly(VAF-co-VSC-co-VGA) by dynamic light scattering. | ||
2.3. Cellular uptake of nanoparticles
We used CLSM to investigate the cellular uptake of galactose-functionalized nanoparticles self-assembled from poly(VAF-co-VSC-co-VGA), poly(VAF-co-VGA) and poly(VSC-co-VGA) by HepG2 human hepatoma cells, which overexpressed galactose-binding asialoglycoprotein receptors. The aggregates without galactose from poly(VAF-co-VSC), poly(VAF) and poly(VSC) were used as controls. RPMI 1640 media containing FITC-labeled of these six kinds of aggregates respectively were used to culture HepG2 human hepatoma cells for 3 h. Then the samples were visualized by CLSM at an excitation wavelength of 488 nm and an emission wavelength of 493–561 nm. Obvious fluorescence intensity was observed in the cytosol and nucleus regions of HepG2 cells incubated with galactose-functionalized aggregates (Fig. 4a–c), while there appeared to be little fluorescence intensity in the HepG2 cells incubated with aggregates without galactose (Fig. 4d–f). Therefore, the galactose-functionalized aggregates could effectively realize cellular internalization of Ara-C and FUDR. | ||
| Fig. 4 Confocal laser scanning microscopy images of HepG2 cells after incubation in RPMI 1640 medium containing (a) FITC-labeled poly(VAF-co-VSC-co-VGA), (b) FITC-labeled poly(VAF-co-VGA), (c) FITC-labeled poly(VSC-co-VGA), (d) FITC-labeled poly(VAF-co-VSC), (e) FITC-labeled poly(VAF) and (f) FITC-labeled poly(VSC) aggregates for 3 h. | ||
2.4. Cell cytotoxicity and hepatoma targeting ability of nanoparticles
The most meaningful aspects in our study were the potential of synergistic cytotoxic effect of the drugs (Ara-C and FUDR) and the hepatoma-targeting function of the micelles. Cell cytotoxicity of the resulting galactose-functionalized nanoparticles from poly(VAF-co-VSC-co-VGA), poly(VAF-co-VGA) and poly(VSC-co-VGA) was investigated toward HepG2 human hepatoma cells. HepG2 cells were exposed to the nanoparticles, free Ara-C, free FUDR and free Ara-C plus free FUDR in various concentrations, respectively. The cell cytotoxicity effects were determined by MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) dye reduction assays. In comparison, nanoparticles self-assembled from the galactose-functionalized random copolymer poly(VAF-co-VSC-co-VGA) could significantly inhibit the growth of HepG2 human hepatoma cells compared with the physically mixed aggregates of poly(VSC-co-VGA) and poly(VAF-co-VGA) or the mixture of free Ara-C and free FUDR (Fig. 5a). The inhibition increased with the increase of the nanoparticle concentration. Also, single-drug-containing aggregates of poly(VSC-co-VGA) and poly(VAF-co-VGA) had a little more effective growth-inhibitory activity than Ara-C and FUDR small molecules (Fig. 5b). We conjectured that the combination of the hepatoma-targeting function and the potential synergistic effect enhanced the growth-inhibitory activity of the novel nanoparticles toward HepG2 cells, which overexpressed galactose-binding asialoglycoprotein receptor (ASGPR). In addition, the simultaneous release of Ara-C and FUDR made the synergistic effect more obvious as poly(VAF-co-VSC-co-VGA) aggregates showed higher cytotoxic effects than a mixture of poly(VSC-co-VGA) and poly(VAF-co-VGA) aggregates as well as the mixture of FUDR and Ara-C. The research of Younsoo Bae and colleagues revealed similar results.24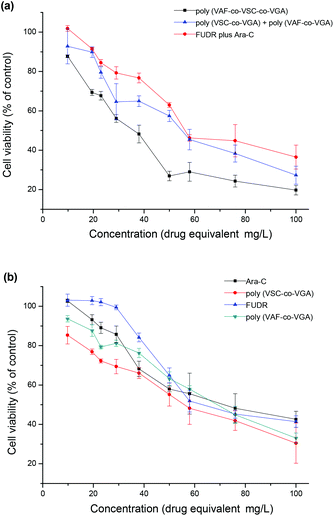 | ||
| Fig. 5 Cytotoxicities of different samples against HepG2 cells. HepG2 cells were, respectively, incubated with the drug small molecules poly(VSC-co-VGA), poly(VAF-co-VGA) and their mixture, poly(VAF-co-VSC-co-VGA) aggregates for 24 h. (a) Comparison between poly(VAF-co-VSC-co-VSG), poly(VSC-co-VSG) plus poly(VAF-co-VSG) and FUDR plus Ara-C. (b) Comparison between single drug-containing polymer aggregates and their corresponding drug small molecules. Data are shown as mean ± SD (n = 4). | ||
To confirm our hypothesis, the inhibition assay of free D-galactose in HepG2 cells was carried out. HepG2 cells were exposed to various concentrations (0–100 μg mL−1) of D-galactose for 1 h. Then the HepG2 cells were incubated in the serum-free RPMI 1640 medium with or without the multidrug-conjugating nanoparticles (150 μg mL−1) for another 24 h. The result is shown in Fig. 6. The growth-inhibitory activity of the multidrug-containing nanoparticles decreased with increasing the concentration of free galactose. The cytotoxicity of the nanoparticles toward HepG2 cells was almost inhibited while the galactose concentration was 100 μg mL−1. This suggested that asialoglycoprotein receptor-mediated uptake by galactose ligands on the surface of micelles was involved in the cytotoxicity of the nanoparticles.
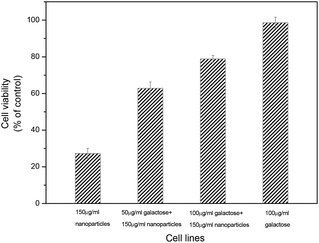 | ||
| Fig. 6 Inhibition assay of cytotoxicity of poly(VAF-co-VSC-co-VGA) in HepG2 cells with small-molecule D-galactose. HepG2 cells were incubated with poly(VAF-co-VSC-co-VGA) (150 μg mL−1) for 24 h after being exposed to D-galactose with various concentrations (0, 50, 100 μg mL−1) for 1 h. The well without poly(VAF-co-VSC-co-VGA) was chosen as a control. The cytotoxicity was measured by a MTT assay. Data are shown as mean ± SD (n = 4). | ||
Therefore, the galactose-functionalized poly(VAF-co-VSC-co-VGA) aggregates had an evident hepatoma-targeting function and a potential synergistic effect, yielding efficient cellular uptake and multi-drug release in HepG2 cells, and thus enhanced anti-tumorous cell cytotoxicity.
2.5. In vitro release of drugs
We carried out the in vitro release experiments of Ara-C and FUDR in pH = 7.4 phosphate buffer solution (PBS, simulated extracellular environment) and pH = 5.8 phosphate buffer solution (PBS, simulated intracellular environment of HepG2 cells) via dialysis experiments, to see whether both drugs could be slowly released from the nanoparticles of galactose-functionalized poly(VAF-co-VSC-co-VGA). The release process of Ara-C and FUDR was determined by HPLC, and the results are shown in Fig. 7 and 8. From the figures, we can see that both drugs conjugated to the copolymer were released from the micelles slowly but enduringly, and Ara-C was released relatively faster than FUDR. The cumulative released amounts of Ara-C and FUDR after 144 h (6 days) were 48% and 13% in pH 7.4 PBS and 37% and 10% in pH 5.8 PBS. We can see that the release of drugs from nanoparticles in pH 7.4 PBS was a little faster than that in pH 5.8 PBS. This was probably because that the amino group of Ara-C could slightly increase the alkalescence in pH 7.4 PBS. In addition, a lower pH would make the amino groups on the surface of the nanoparticles protonated, thus inducing an electrostatic shield effect. Then H+ in the solution would be prevented from approaching the ester bond inside the nanoparticles, which affected the hydrolysis of the nanoparticles. The different release rate of Ara-C and FUDR was probably due to the different protonation ability and solubility of these two molecules. This in vitro release behavior indicated that the micellar nanoparticles could be used as a novel multidrug delivery system.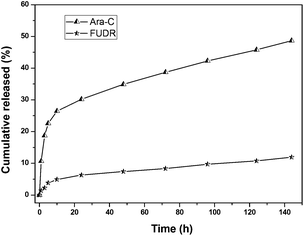 | ||
| Fig. 7 In vitro release of nanoparticles formed from poly(VAF-co-VSC-co-VGA) in pH 7.4 phosphate buffer solution at 37 °C. The concentrations of drug released were determined by HPLC with UV detection at 270 nm. | ||
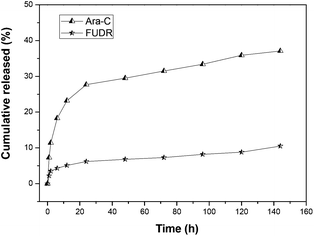 | ||
| Fig. 8 In vitro release of nanoparticles formed from poly(VAF-co-VSC-co-VGA) in pH 5.8 phosphate buffer solution at 37 °C. The concentrations of drug released were determined by HPLC with UV detection at 270 nm. | ||
3. Conclusion
A novel multidrug-conjugating hepatoma-targeting amphiphilic random copolymer was prepared by combining enzymatic transesterifications and radical polymerization. The polymeric micelles assembled from the copolymer were regular spheres with a hydrodynamic diameter of 472 ± 45 nm in aqueous solution. The low CAC value (1 mg L−1) allowed their use in very dilute aqueous media such as body fluids. Drug release experiments revealed that both drugs incorporated in the nanoparticles could be released slowly and enduringly, which confirmed that these novel nanoparticles could deliver multidrug agents to tumors simultaneously. Most interestingly, the cellular uptake assay and cell cytotoxicity tests showed that the polymeric micelles had an evident targeting function to HepG2 human hepatoma cells and showed enhanced cytotoxic activity by concurrently delivering Ara-C and FUDR, probably attributed to the synergistic action.In conclusion, this novel galactose-functionalized amphiphilic random copolymer is promising for delivering two different chemotherapeutic agents to target tumors simultaneously. Further studies about the in vivo disposition of the aggregates are being conducted in our laboratory.
4. Experimental section
4.1. Materials
α,α′-Azobis-(isobutyronitrile) (AIBN) was purchased from Fluka and purified by recrystallization in methanol and dried at room temperature under vacuum. 5′-O-Vinylsebacoyl-cytarabine (VSC) and 3′-O-vinyladipoyl-fluorodeoxyuridine (VAF) were synthesized and purified according to the literature.35 6-O-Vinylsebacoyl-D-galactose (VGA) was prepared and purified as described in the paper.36 Roswell Park Memorial Institute (RPMI) 1640 incubation medium and fetal calf serum were purchased from Genom BioMed Technology Co. Ltd (Hangzhou, People's Republic of China). N,N-Dimethylformamide (DMF) was HPLC grade. Dimethyl sulfoxide, tetrahydrofuran (THF), methanol, acetone, and all other chemicals were of appropriate analytical grade.4.2. Measurements
1H NMR spectra were measured by a Bruker AMX-500 MHz FT-NMR spectrometer using DMSO-d6 as the solvent and tetramethylsilane (TMS) as an internal reference. FTIR spectra were recorded on a Nicolet Nexus 670 FTIR spectrophotometer at room temperature in the range 4000–500 cm−1. Polymer molecular weights were measured by gel permeation chromatography (GPC) equipped with a refractive-index detector (Waters 2410) and Waters Styragel® HR columns, and DMF was used as the mobile phase. The morphology of the nanoparticles formed from the copolymer was investigated by transmission electron microscopy (TEM) on a JEM-1230 transmission electron microscope at an accelerating voltage of 80 kV. A drop of the aqueous solution of nanoparticles was placed on the carbon-coated copper grid and dried in air at room temperature. Dynamic light scattering (DLS) was carried out on a Nanoseries (Malvern, UK) zetasizer with 90° collecting optics to determine the hydrodynamic size (DH) and the size distribution (PDI) of the nanoparticles. The sample solution was treated with a 0.45 μm pore size filter and the measurements were carried out at 25 °C.4.3. Synthesis of poly(3′-O-vinyladipoyl-fluorodeoxyuridine-co-5′-O-vinylsebacoyl-cytarabine-co-6-O-vinylsebacoyl-galactose) (poly(VAF-co-VSC-co-VGA))
Poly(3′-O-vinyladipoyl-fluorodeoxyuridine-co-5′-O-vinylsebacoyl-cytarabine-co-6-O-vinylsebacoyl-galactose) (poly(VAF-co-VSC-co-VGA)) was prepared by adding VAF (200 mg, 0.5 mmol), VSC (225 mg, 0.5 mmol), VGA (195 mg, 0.5 mmol) into a sealed flask containing DMF (0.60 mL) and AIBN (30 mg). The mixture was vacuumed by pump, and then stirred under nitrogen at 70 °C for 24 h. The resulting product was precipitated in acetone and washed with DMF repeatedly to remove the residual monomers and then dried under vacuum to obtain light yellow poly(VAF-co-VSC-co-VGA) (284 mg, 45.8%). Mn = 4.34 × 104, Mw/Mn = 1.41. IR (KBr): v (cm−1) 3435, 1736, 1647, 1260, 1078. 1H-NMR (500 MHz, DMSO-d6, δ): 11.98 (1H, NH of fluorodeoxyuridine), 8.07 (1H, 6-H of fluorodeoxyuridine), 7.56 (1H, 6-H of cytarabine), 7.27 (1H, –NH2 of cytarabine), 7.12 (1H, –NH2 of cytarabine), 6.66, 6.31 (1-OH of D-galactose), 6.15 (1H, 1′-H of fluorodeoxyuridine), 5.76–5.64 (1′-H, 5-H, 2′-OH, 3′-OH of cytarabine; 5′-OH of fluorodeoxyuridine), 5.30–2.85 (–CH–O–; 1-H, 2-H, 3-H, 4-H, 5-H, 6-H, 2-OH, 3-OH and 4-OH of D-galactose; 1H, 3′-H, 4′-H, 5′-H of fluorodeoxyuridine; 2′-H, 4′-H, 3′-H, 5′-H, 5′-H of cytarabine), 2.161 (2′-H of fluorodeoxyuridine; 8H, O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–CH2), 1.58 (8H, –CH2–), 1.30–1.38 (8H, –CH2–; 4H, –CH2– of backbone of the polymer).
C–CH2), 1.58 (8H, –CH2–), 1.30–1.38 (8H, –CH2–; 4H, –CH2– of backbone of the polymer).
4.4. Synthesis of poly(3′-O-vinyladipoyl-fluorodeoxyuridine-co-6-O-vinylsebacoyl-galactose) (poly(VAF-co-VGA)) and poly(5′-O-vinylsebacoyl-cytarabine-co-6-O-vinylsebacoyl-galactose) (poly(VSC-co-VGA))
VAF (200 mg, 0.5 mmol) and VSC (225 mg, 0.5 mmol) were respectively dissolved in a sealed flask containing VGA (195 mg, 0.5 mmol) and DMSO (0.4 mL). AIBN (20.0 mg) was then added as an initiator. The mixture was degassed by pump and stirred under nitrogen atmosphere at 70 °C for 24 h. The resulting products were precipitated in acetone and washed with methanol repeatedly to remove the residual monomers, and then dried under vacuum to obtain light yellow products poly(VAF-co-VGA) (163 mg, 41.3%). Mn = 2.32 × 104, Mw/Mn = 1.73. IR (KBr): v (cm−1) 3449, 3240, 1716, 1258, 1095. 1H-NMR (400 MHz, DMSO-d6, δ): 11.80 (1H, NH of fluorodeoxyuridine), 8.18 (1H, 6-H of fluorodeoxyuridine), 6.65, 6.35 (1-OH of D-galactose), 6.12 (1H, 1′-H of fluorodeoxyuridine), 5.36–3.06 (5′-OH, 3′-H, 4′-H, 5′-H of fluorodeoxyuridine; 1-H, 2-H, 3-H, 4-H, 5-H, 6-H, 2-OH, 3-OH and 4-OH of D-galactose; –CH–O–), 2.25 (2′-H of fluorodeoxyuridine; OC–CH2), 1.68–1.23 (–CH2–), and poly (VSC-co-VGA) (159 mg, 52.3%). Mn = 1.97 × 104, Mw/Mn = 1.56. IR (KBr): v (cm−1) 3350, 3228, 1736, 1647, 1286, 1082. 1H-NMR (400 MHz; DMSO-d6; δ): 7.47 (1H, 6-H of cytarabine), 7.18 (1H, –NH2 of cytarabine), 7.09 (1H, –NH2 of cytarabine), 6.66, 6.35 (1-OH of D-galactose), 6.08 (1H, 1′-H of cytarabine), 5.69–3.05 (1′-H, 5-H, 2′-OH, 3′-OH, 2′-H, 4′-H, 3′-H, 5′-H, 5′-H of cytarabine; 1-H, 2-H, 3-H, 4-H, 5-H, 6-H, 2-OH, 3-OH and 4-OH of D-galactose; –CH–O–), 2.30 (2H, OC–CH2–), 1.48–1.18 (–CH2–).
4.5. Fluorescence measurements
The micellization ability of the resulting amphiphilic random copolymer was proved by fluorescence spectroscopy using pyrene as the hydrophobic fluorescent probe. Five samples with the copolymer aggregate concentrations ranging from 1 × 10−5 to 0.1 g L−1 were prepared with a constant pyrene concentration of 6 × 10−7 M. The samples were incubated for 2 h at 50 °C and cooled for 12 h at room temperature. Then the fluorescence spectra were recorded on a Shimadzu RF-5301 PC spectrofluorophotometer. The excitation wavelength was 339 nm and the emission spectra were recorded from 360 to 460 nm. The excitation and emission bandwidths were 5 nm and 3 nm respectively. The value of the emission intensity ratio of the third band (I384) to the first band (I373) was analyzed as a function of the copolymer concentration. The critical micellar concentration (CMC) value of the copolymer was determined from the crossover point of the horizontal tangent with nearly aptotic values at low concentrations and the diagonal line with rapid increased intensity ratio.4.6. Micelles preparation
The micelles self-assembled from the galactose-functionalized amphiphilic random copolymer with a dialysis method. Poly(VAF-co-VSC-co-VGA) was dissolved in DMSO at an initial concentration of 0.1 wt% at room temperature. Then ultrapure water was added dropwise to the solution under stirring until the final concentration of water was 25 wt%. The resulting mixture was dialyzed using a dialysis membrane (MWCO = 3500) against ultrapure water for 2 days to remove DMSO from the solution and was finally lyophilized. Nanoparticles self-assembled from poly(VAF-co-VGA) and poly(VSC-co-VGA) were prepared in just the same way.4.7. Cellular uptake assay
In order to confirm whether the galactose-functionalized nanoparticles self-assembled from poly(VAF-co-VSC-co-VGA), poly(VAF-co-VGA) and poly(VSC-co-VGA) had a targeting function against liver carcinoma cells, a cellular uptake assay of the nanoparticles was carried out using HepG2 human hepatoma cells. Nanoparticles from galactose-free polymers poly(VAF-co-VSC), poly(VAF) and poly(VSC) were chosen as controls. First, the polymers were respectively dissolved in pH 9.6 borate saline buffer solution (0.2 mM drugs equivalent concentration). Then 10 μL DMSO solution of FITC (1% w/v) was added to 5 mL resulting solutions. Following stirring for 12 h at 25 °C, The FITC-labeled samples were respectively dialyzed (MWCO = 3500 Da) for 72 h against pH 8.0 phosphate buffer solution (PBS). The prepared HepG2 cells were incubated in RPMI 1640 media containing FITC-labeled samples (0.04 mM drugs equivalent concentration) for 3 h at 37 °C. Then the media were removed and the HepG2 cells were washed three times using PBS to remove unbound samples. Lastly, FITC fluorescence was observed by CLSM (LSM 710nlo, Zeiss, Germany).4.8. Cell cytotoxicity assay
HepG2 human hepatoma cells were chosen as target cells to verify the antitumor activity of the multidrug-conjugating nanoparticles based on the galactose-functionalized amphiphilic random copolymer. The HepG2 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium with 10% fetal calf serum and the cell density was adjusted to 2.5 × 104 cells per mL. 200 μL aliquots of the suspension were added to the wells of the sterile 96-well plate and the plate was placed in the cell culture incubator at 37 °C under 5% CO2 for 16 h. Then the medium was replaced with 200 μL serum-free RPMI 1640 medium containing various concentrations (100, 76, 58, 50, 38, 29, 23, 19, 9.7 mg L−1 Ara-C equivalent concentration; 100, 80, 58, 50, 40, 29, 24, 20, 9.8 mg L−1 FUDR equivalent concentration, respectively) of the samples (free Ara-C, free FUDR, free Ara-C plus FUDR, nanoparticles of poly (VAF-co-VSC-co-VGA), poly (VAF-co-VGA), poly (VSC-co-VGA), and poly (VAF-co-VGA) plus poly (VSC-co-VGA)) and then further incubated for 24 h. The cytotoxic effects of the samples were evaluated by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) dye reduction assay. Moreover, the inhibition assay of small molecule D-galactose in HepG2 cells was carried out. HepG2 cells were exposed to various concentrations (0–100 μg mL−1) of D-galactose for 1 h before incubation in the serum-free RPMI 1640 medium with or without the multidrug-conjugating nanoparticles (150 μg mL−1) for another 24 h. The viability of cells was detected with a microplate reader and was expressed as a percentage of the viability ratio of treated cells to the control cells.4.9. In vitro release experiment
The in vitro release behavior of drugs from the micelles was studied by a dialysis experiment. Nanoparticles (1.0 mg) self-assembled from poly(VAF-co-VSC-co-VGA) were added into 1.00 mL pH 7.4, 0.2 M phosphate buffer solution and pH 5.8, 0.2 M phosphate buffer solution respectively. The resulting solution was added into a dialysis bag (MWCO = 3500) and then immersed into an erlenmeyer flask with 5.00 mL corresponding buffer solutions. The erlenmeyer flask was shaken under 100 rpm at 37 °C. Periodically, the whole buffer (5.00 mL) taken out from the incubation erlenmeyer flask and replaced by another 5 mL fresh buffer. Finally, the concentrations of drugs released from the nanoparticles were determined by HPLC.Acknowledgements
Financial support by the National Natural Science Foundation of China (Nos. 20704037, 20874086) is gratefully acknowledged.Notes and references
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347–360 CrossRef CAS
.
- R. Satchi-Fainaro, R. Duncan and C. M. Barnes, Adv. Polym. Sci., 2006, 193, 1–65 CrossRef CAS
- N. Nishiyama and K. Kataoka, Pharmacol. Ther., 2006, 112, 630–648 CrossRef CAS
- R. Duncan, Nat. Rev. Cancer, 2006, 6, 688–701 CrossRef CAS
- A. Lavasanifar, J. Samuel and G. S. Kwon, Adv. Drug Delivery Rev., 2002, 54, 169–190 CrossRef CAS
- R. Haag, Angew. Chem., Int. Ed., 2004, 43, 278–282 CrossRef CAS
- M. J. Vicent, F. Greco, R. I. Nicholson, A. Paul, P. C. Griffiths and R. Duncan, Angew. Chem., Int. Ed., 2005, 44, 4061–4066 CrossRef CAS
- Z. G. Xie, H. L. Guan, X. S. Chen, C. H. Lu, L. Chen, X. L. Hu, Q. Shi and X. B. Jing, J. Controlled Release, 2007, 117, 210–216 CrossRef CAS
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS
- H. Maeda, T. Sawa and T. Konno, J. Controlled Release, 2001, 74, 47–61 CrossRef CAS
- N. Nasongkla, E. Bey, J. M. Ren, H. Ai, C. Khemtong, J. Setti Guthi, S. F. Chin, A. D. Sherry, D. A. Boothman and J. M. Gao, Nano Lett., 2006, 6, 2427–2430 CrossRef CAS
- P. A. McCarron, W. M. Marouf, D. J. Quinn, F. Fay, R. E. Burden, S. A. Olwill and C. J. Scott, Bioconjugate Chem., 2008, 19, 1561–1569 CrossRef CAS
- A. H. Lin, Y. M. Liu, Y. Huang, J. B. Sun, Z. F. Wu, X. Zhang and Q. N. Ping, Int. J. Pharm., 2008, 359, 247–253 CrossRef CAS
- D. Kim, E. S. Lee, K. T. Oh, Z. G. Gao and Y. H. Bae, Small, 2008, 4, 2043–2050 CrossRef CAS
- M. R. Kano, Y. Bae, C. Iwata, Y. Morishita, M. Yashiro, M. Oka, T. Fujii, A. Komuro, K. Kiyono, M. Kaminishi, K. Hirakawa, Y. Ouchi, N. Nishiyama, K. Kataoka and K. Miyazono, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3460–3465 CrossRef CAS
- G. Ashwell and J. Harford, Annu. Rev. Biochem., 1982, 51, 531–554 CrossRef CAS
- E. Jubelt, L. Moine and G. Barratt, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3178–3187 CrossRef
- C. Y. Wu, J. Quan, J. G. Xie, C. Branford-White, L. M. Zhu, Y. Yu and Y. J. Wang, Polym. Bull., 2011, 67, 593–608 CrossRef CAS
- S. Bhatia, A. Mohr, D. Mathur, V. S. Parmar, R. Haag and A. Prasad, Biomacromolecules, 2011, 12, 3487–3498 CrossRef CAS
- A. David, P. Kopeckova, A. Rubinstein and J. Kopecek, Bioconjugate Chem., 2001, 12, 890–899 CrossRef CAS
- L. Ying, C. Yin, R. X. Zhuo, K. W. Leong, H. Q. Mao, E. T. Kang and K. G. Neoh, Biomacromolecules, 2003, 4, 157–165 CrossRef CAS
- F. L. Mi, Y. Y. Wu, Y. L. Chiu, M. F. Chen, H. W. Sung, S. H. Yu, S. S. Shyu and M. F. Huang, Biomacromolecules, 2007, 8, 892–898 CrossRef CAS
- F. Ahmeda, R. I. Pakunlu, A. Brannan, F. Bates, T. Minkob and D. E. Discher, J. Controlled Release, 2006, 116, 150–158 CrossRef
- Y. Bae, T. A. Diezi, A. Zhao and G. S. Kwon, J. Controlled Release, 2007, 122, 324–330 CrossRef CAS
- T. Lammers, V. Subr, K. Ulbrich, P. Peschke, P. E. Huber, W. E. Hennink and G. Storm, Biomaterials, 2009, 30, 3466–3475 CrossRef CAS
- P. J. Shamia, A. E. Maciagb, J. K. Eddingtona, V. Udupia, K. M. Kosaka, J. E. Saavedrab and L. K. Keefer, Leuk. Res., 2009, 33, 1525–1529 CrossRef
- F. Greco, M. J. Vicent, N. A. Penning, R. I. Nicholson and R. Duncan, J. Drug Targeting, 2005, 13, 459–470 CrossRef CAS
- F. Greco, M. J. Vicent, S. Gee, A. T. Jones, J. Gee, R. I. Nicholson and R. Duncan, J. Controlled Release, 2007, 117, 28–39 CrossRef CAS
- K. Breist, J. Balzarini, M. L. Sandvold, F. Myhren, M. Martinsen, E. D. Clercq and Ø. Fodstad, Cancer Res., 1999, 59, 2944–2949 Search PubMed
- B. Daniel, D. Longley, P. Harkin and P. G. Johnston, Nat. Rev. Cancer, 2003, 3, 330–338 CrossRef
- T. B. Cai, X. P. Tang, J. Nagorski, P. G. Brauschweiger and P. G. Wang, Bioorg. Med. Chem., 2003, 11, 4971–4975 CrossRef CAS
- H. Schott, S. Schott and R. A. Schwendener, Bioorg. Med. Chem., 2009, 17, 6824–6831 CrossRef CAS
- F. M. Menger and M. J. Rourk, J. Org. Chem., 1997, 62, 9083–9088 CrossRef CAS
- I. V. Bijnsdorp, R. A. Schwendener, H. Schott, I. Fichtner, K. Smid, S. Schott, A. C. Laan and G. J. Peters, Nucleosides, Nucleotides Nucleic Acids, 2007, 26, 1619–1624 CAS
- C. Yin, X. Li, Q. Wu, J. L. Wang and X. F. Lin, J. Colloid Interface Sci., 2010, 349, 153–158 CrossRef CAS
- Q. Wu, N. Wang, Y. M. Xiao, D. S. Lu and X. F. Lin, Carbohydr. Res., 2004, 339, 2059–2067 CrossRef CAS
- N. Wang, Q. Wu, Z. C. Chen, D. S. Lu and X. F. Lin, Bioorg. Med. Chem. Lett., 2005, 15, 4064–4067 CrossRef CAS
- A. H. Xia, Q. Wu, B. K. Liu and X. F. Lin, Enzyme Microb. Technol., 2008, 42, 414–420 CrossRef CAS
- Y. L. Zheng, L. Zhong, W. Huang, Y. F. Zhou and D. Y. Yan, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4428–4438 CrossRef CAS
- W. Su, X. H. Luo, H. F. Wang, L. Li, J. Feng, X. Z. Zhang and R. X. Zhuo, Macromol.
Rapid Commun., 2011, 32, 390–396 CrossRef CAS
- H. Y. Hong, Y. Y. Mai, Y. F. Zhou, D. Y. Yan and J. Cui, Macromol. Rapid Commun., 2007, 28, 591–596 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: The 1H-NMR spectra of monomers and copolymer. See DOI: 10.1039/c3bm60027k |
| This journal is © The Royal Society of Chemistry 2013 |