Hyaluronan and self-assembling peptides as building blocks to reconstruct the extracellular environment in skin tissue†
Daniela S.
Ferreira
ab,
Alexandra P.
Marques
ab,
Rui L.
Reis
ab and
Helena S.
Azevedo
*ab
a3B's Research Group – Biomaterials, Biodegradables and Biomimetics, University of Minho, Headquarters of the European Institute of Excellence on Tissue Engineering and Regenerative Medicine, AvePark, 4806-909 Taipas, Guimarães, Portugal. E-mail: hazevedo@dep.uminho.pt; Fax: +351 253 510 909; Tel: +351 253 510 907
bICVS/3B's – PT Government Associate Laboratory, Braga/Guimarães, Portugal
First published on 6th June 2013
Abstract
Self-assembling bioactive membranes, incorporating hyaluronan, a structural component of the skin extracellular matrix (ECM), and peptide amphiphiles presenting biochemical signals, are proposed in this work for recapitulating some aspects of the skin tissue microenvironment. In the herein presented strategy, the availability of cell-adhesion ligands (0–50% RGDS epitope) within 2D membranes is controlled aiming at mastering the adhesion of human dermal fibroblasts under serum-free culture conditions. The membranes were characterized with respect to their microstructure, by scanning electron microscopy (SEM), epitope distribution, degradability and cell behavior, regarding adhesion, proliferation and cytoskeleton organization. SEM of the membrane surface showed a network of nanofibers that are remarkably reminiscent of the filamentous structure found in the ECM. Confocal microscopy images, using a fluorescently labeled RGDS-peptide, showed that the RGDS signal is uniformly distributed on the membranes. Degradation studies indicated that the membranes are susceptible to enzymatic degradation by hyaluronidase. In the presence of the enzyme at physiological concentration, the membranes degrade gradually over time. When grown on membranes with the cell recognition epitope RGDS, fibroblasts had spread out and elongated, exhibiting extended filopodia interacting with fibrillar structure of the membrane surface, thus showing improved adhesion to the substrate. This study demonstrates the positive effect of the RGDS epitope, presented on a self-assembled membrane, in promoting cell–matrix interactions.
1. Introduction
The extracellular matrix (ECM) of tissues is a dynamic and hierarchically organized composite structure of various fibrillar proteins and glycosaminoglycans. This network not only has a structural role, providing support and tensile strength for tissues and acting as scaffolds for cell adhesion and organization, but also serves as a storage site for growth factors, chemokines and cytokines, and as a template for tissue morphogenesis and cell differentiation.1–3In skin, the ECM is the largest component of the dermal layer, being composed of structural proteins, like elastin, that confers skin elasticity, and collagens, primarily type I and III, which provide structure, strength and integrity.4 Cell-adhesive proteins, like fibronectin, laminin and vitronectin are also present in skin ECM. These glycoproteins have the capacity to bind to cells, via integrins, and to other components of the ECM, namely to glycosaminoglycans, except hyaluronan (HA), which is one of the major ECM components in skin.4,5 HA is an extremely large polymer made up of N-acetylglucosamine and glucuronic acid disaccharide repeating units (Fig. 1A). High molecular weight HA acts as an ECM organizer which concentrates and organizes the assembly of other proteins in the ECM by providing a macromolecular template, thus contributing to the tissue architecture and function during homeostasis.6,7 These properties confer on HA many unique advantages as a starting material for skin regeneration applications.
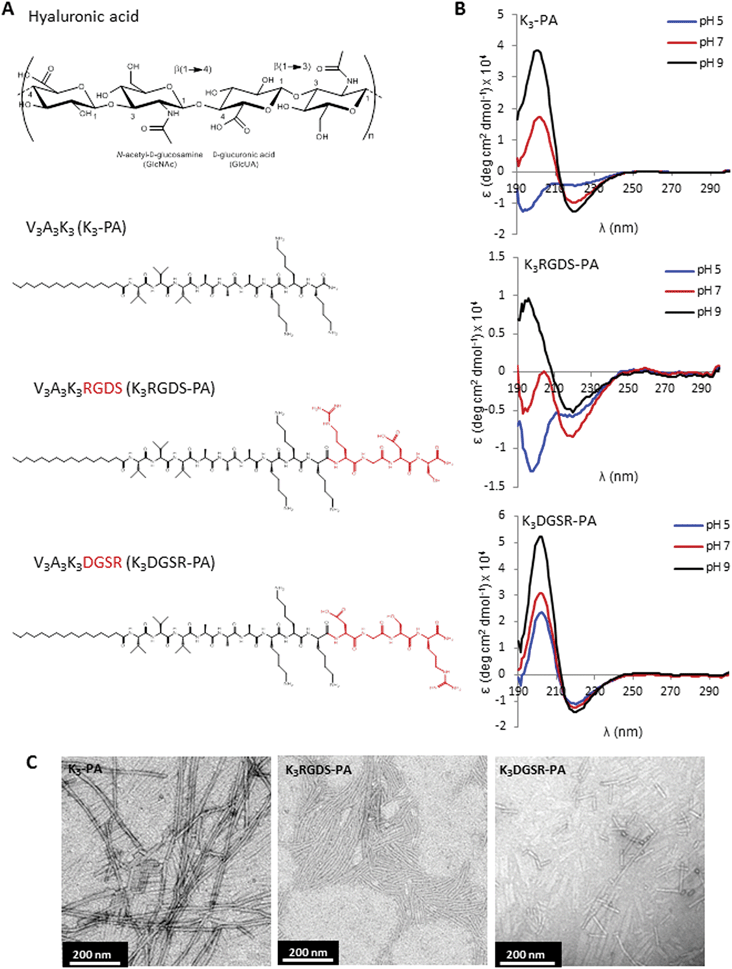 | ||
| Fig. 1 Peptide design and characterization. (A) Chemical structure of the building blocks used for preparing the self-assembled membranes, hyaluronan (HA) and different peptide amphiphiles (PAs): V3A3K3 containing positively charged lysines (KKK) to bind the anionic HA, one containing the RGDS epitope (K3RGDS-PA), a scrambled version (K3DGSR-PA). (B) Circular dichroism spectra of peptide solutions (3.3 × 10−5 M) at pH 5, 7 and 9 showing a predominantly β-sheet secondary structure. (C) TEM images of PA nanostructures formed by the deposition of 0.1 mM solutions in water followed by air drying on a carbon-coated TEM grid. | ||
Cell adhesion to native ECM is mediated through the binding of integrin proteins on the cell surface and specific epitopes present in proteins of the ECM, creating a focal adhesion, responsible for anchoring the cell and the communication between the cell cytoskeleton and the surrounding environment.8 One of the ECM cell adhesive proteins, fibronectin, binds to integrins through a domain containing Arg-Gly-Asp-Ser (RGDS).9 This sequence, first proposed by Pierschbacher and Rouslahti, functions as a general cell adhesive sequence10 and has been widely used in biomaterials functionalization, including HA-based hydrogels,11–13 improving cell adhesion and subsequent proliferation.14–17
Peptides represent an interesting family of building blocks which can self-assemble to create a large number of nanostructures, such as micelles, vesicles and nanofibers.18–20 Thus, by self-assembly, structurally simple building blocks can be easily gathered to create functionally complex materials.8
Bottom-up approaches based on the self-assembly of small molecules provide a unique set of advantages to create biomaterials, as they offer the possibility of controlling the architecture, shape and dimensions of the bioactive nanostructures, as well as the spatial display and density of the bioactive signals.21 The architectural resemblance of self-assembled nanofibers to filamentous structures found in natural ECMs represents an additional feature to attain superior biomimetic scaffolds, and a clear advantage in biomaterials engineering.
The fabrication of artificial ECMs can be used to mimic the properties of native tissues as well as to reconstruct cellular microenvironments in vitro. We address this challenge by combining self-assembling peptides (peptide amphiphiles) integrating biochemical signals (RGDS ligand) to permit cell adhesion and spreading, and functional molecules (HA), as components of our matrix. These components were shown to self-assemble in 2D membranes.22–24 Through self-assembly, epitope spatial organization can be controlled at the micrometer and nanometer scales to guide cellular behavior. Materials that selectively interact with cells may be helpful in improving our understanding of key structural and biochemical ECM components, and ultimately, harnessing the presentation of specific cues to cells. This represents a simplified approach to deconstruct the skin niche and to identify the effect of individual niche components over cell behavior.
2. Materials and methods
Hyaluronan (HA) and fluorescein HA
The hyaluronan used in all experiments had an average molar mass of 2 MDa and was purchased from Lifecore Biomedical, Inc. (Chaska, USA). HA was fluorescently labeled with fluoresceinamine (Fig. 3B) using N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) chemistry and following the procedure described by Gajewiak et al.25 Briefly, HA (50 mg) was dissolved in 20 mL of water to give a 0.25% (w/v) solution, which was then mixed with a solution of 5 mg of fluoresceinamine (Sigma, USA) in 20 mL of dimethylformamide. Next, 100 mg of N-hydroxysuccinimide (NHS, Sigma, USA) was added, and the solution pH was adjusted to 4.75 with 0.01 M HCl. Finally, 50 mg of EDC (Sigma, USA) was added maintaining the solution pH at 4.75. After 12 h, the solution was transferred to dialysis tubing (2000 Da MWCO, Sigma, USA) and dialyzed exhaustively against 100 mM NaCl, followed by dialysis against distilled water and lyophilization.Peptide amphiphiles synthesis and purification
Three different peptide amphiphiles (PAs) were synthesized in this work, consisting of a peptide segment covalently linked to a 16-carbon alkyl chain: C15H31CO-V3A3K3 (K3-PA, filler), C15H31CO-V3A3K3RGDS (K3RGDS-PA) and C15H31CO-V3A3K3DGSR (K3DGSR-PA, scrambled) (Fig. 1). The peptides were synthesized in a CS Bio 136XT automated peptide synthesizer (CS Bio, USA) using standard 9-fluorenylmethoxycarbonyl (Fmoc) based solid phase chemistry and a 4-methylbenzhydrylamine (MBHA) rink amide resin. Amino acid couplings were performed using 4 equivalents (4 mmol) of Fmoc protected amino acids (Novabiochem®), 4 equivalents of O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU, Novabiochem®) and 6 equivalents of N,N-diisopropylethylamine (DIEA, Sigma, USA). Fmoc deprotections were performed with 20% piperidine (Sigma, USA) in dimethylformamide. A palmitic acid (C16H32O2, Calbiochem, USA) tail was manually coupled under the same conditions as the Fmoc-amino acids. Peptide cleavage from the resin and removal of the protecting groups were carried out in a mixture of trifluoroacetic acid (TFA, Sigma, USA)/triisopropylsilane (TIS, Alfa Aesar)/water (95/2.5/2.5) for 3 h at room temperature. The peptide mixture was collected and excess TFA was removed by rotary evaporation. The resulting viscous peptide solution was triturated with cold diethyl ether. The white precipitate was collected by filtration, washed with cold ether, and allowed to dry under vacuum overnight. The peptide mass was confirmed by electrospray ionization mass spectrometry (ESI-MS, Thermo Electron Corporation Finnigan LXQ MS Waltham, USA). Peptides were then purified on a Waters 2545 Binary Gradient high-performance liquid chromatography (HPLC) system using a preparative reverse-phase C18 column (Atlantis Prep OBD T3 column, Waters, USA) and a water/acetonitrile (0.1% TFA) gradient. TFA counter-ions were exchanged by sublimation from 0.01 M hydrochloric acid. Finally, the peptides were dialyzed against ultrapure water using 500 MWCO dialysis tubing (Spectrum Labs, The Netherlands), and lyophilized. Confirmation of mass and purity was done by ESI-MS and HPLC (ESI, Fig. S1–S3†).A fluorescent version of K3RGDS-PA, C15H31CO-V3A3K3KrhodRGDS (Fig. 3B), was also synthesized to allow examining the availability/distribution of the RGDS motif within and on the surface of the membranes. For that, an additional lysine residue, with a 4-methyl trityl (Mtt) protecting group in the ε amine of the lysine residue (Fmoc-Lys(Mtt)-OH), was introduced in the sequence to which the rhodamine dye was attached. The peptide was grown on the resin and after coupling the palmitic tail, the Mtt protecting group was selectively removed with a solution of TFA–TIS–DCM (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:92) at room temperature. Resin was incubated with a deprotection solution for 5 minutes and washed thoroughly with DCM. These steps were repeated until the resin no longer turned yellow. Then, peptide-resin (150 mg) was swollen in 1150 μL of DMF and 50 μL of DIEA. Ten milligrams of 5-(and 6)-carboxytetramethylrhodamine succinimidyl ester (NHS-rhodamine) were dissolved in the supernatant from the beads swelling step and added to the resin. The reaction took place at room temperature, overnight, and protected from light. After repeated washes with DMF and methanol, the peptide was cleaved from the resin by following the procedure described above for the other peptides, and purified by HPLC. Mass was confirmed by matrix assisted laser desorption/ionization mass spectrometry (MALDI-MS, 4800 MALDI-TOF/TOF, AbSciex) (ESI, Fig. S4†).
:4:92) at room temperature. Resin was incubated with a deprotection solution for 5 minutes and washed thoroughly with DCM. These steps were repeated until the resin no longer turned yellow. Then, peptide-resin (150 mg) was swollen in 1150 μL of DMF and 50 μL of DIEA. Ten milligrams of 5-(and 6)-carboxytetramethylrhodamine succinimidyl ester (NHS-rhodamine) were dissolved in the supernatant from the beads swelling step and added to the resin. The reaction took place at room temperature, overnight, and protected from light. After repeated washes with DMF and methanol, the peptide was cleaved from the resin by following the procedure described above for the other peptides, and purified by HPLC. Mass was confirmed by matrix assisted laser desorption/ionization mass spectrometry (MALDI-MS, 4800 MALDI-TOF/TOF, AbSciex) (ESI, Fig. S4†).
Peptide amphiphiles characterization
Characterization of PA–HA interactions
PA–HA membranes preparation
PA–HA membranes were prepared using a 48 well plate as a template in a sterile environment. 150 μL of a 1% (w/v) HA solution was cast on the bottom of the wells and then 150 μL of a 2% (w/v) K3-PA solution was added on top of the HA solution. A membrane is immediately formed upon contact between the two solutions. The membrane was allowed to grow with time (overnight) as reported previously.23 The membranes were rinsed with sterile ultrapure water to ensure the removal of unreacted HA and PA.PA–HA membrane characterization
Cell culture studies
:1000, Sigma-Aldrich, Germany) for 1 min and washed with PBS. Visualization was performed by CLSM (Olympus FluoView 1000, Olympus, Japan). Background was subtracted and images were processed using ImageJ software (NIH, USA).
Data analysis and statistics
Statistical analysis for NAG and DNA quantification was performed using the non-parametric Kruskal–Wallis test, after testing the normality of our data with the Shapiro–Wilk test. Dunn's post-hoc test was carried out to determine the differences between the various conditions in this study. Values of p < 0.05 were considered to determine statistical significant differences between the groups.3. Results and discussion
Peptide design and formulation in the bioactive membranes
The self-assembling peptides used in this study are amphiphilic peptides consisting of a linear hydrophobic segment coupled to a peptide sequence, which includes a β-sheet forming sequence (VVVAAA) and a domain with positively charged amino lysines (KKK) to bind the anionic HA (K3-PA). This class of self-assembling peptides, known as peptide amphiphiles (PAs), was proposed by Stupp's group for different biomedical applications.19,27,28 The fibronectin-derived RGDS epitope was incorporated into the peptide structure (K3RGDS-PA, Fig. 1A) due to its known properties in mediating cell adhesion. Different PA molecules have been co-assembled, allowing for a specific bioactive molecule to be mixed with a non-bioactive diluent molecule29–32 to vary the epitope density on the assembled structures for optimized cell signaling. For example, Webber and co-workers29 have investigated the co-assembly of a positively charged PA containing the RGDS sequence (K3RGDS-PA) with a negative diluent PA (E3-PA) with the aim of producing mixed binary nanofibers. By varying the RGDS composition on surfaces coated with the co-assembled peptide nanofibers, they were able to determine the optimal RGDS density on the adhesion of bone-marrow mononuclear cells. While this co-assembly strategy has been explored by other groups, using coated surfaces29,31 or self-assembled gels,30 the co-assembly of RGDS-containing peptides with HA in self-assembled membranes has been explored by our group.33 In this configuration, the membranes present different densities of biomolecular signals designed to enhance cell adhesion, but other biochemical functionalities can be incorporated, like growth-factor binding sequences relevant to wound healing.Previous studies have shown that the formation of a stable β-sheet is very important and necessary for peptide self-assembly.20,34 Although the formation of β-sheet secondary structure of K3-PA has been demonstrated previously,35 the inclusion of the RGDS (or DGSR) in the PA sequence could disturb that rearrangement and consequently affect the stability of self-assembled membranes. Therefore, circular dichroism (CD) spectroscopy was performed to evaluate the secondary structure of the synthesized peptides (Fig. 1B). The CD analysis of the PAs revealed the presence of hydrogen-bonded structures, namely β-sheets or random coil, depending on the pH. At pH 9, a typical spectrum of a β-sheet structure was observed for all the molecules, with a minimum in the 210–220 nm range, a crossover from positive to negative above 190 nm, and a positive ellipticity around 200 nm. The scrambled peptide showed this conformation in all the studied pH conditions, with stronger peaks at 205 nm. Below pH 9, the K3RGDS-PA showed a random coil conformation. K3-PA presented a β-sheet structure with a minimum at 220 nm and a maximum at 205 nm at pH 7, and a random coil conformation at pH 5. TEM analysis showed the formation of nanofibers for all the peptides, but K3DGSR-PA formed shorter fibers than the K3 and K3RGDS-PAs (Fig. 1C).
PA–HA interaction
Previous studies have shown the ability of K3-PA to interact with HA and form macroscopic structures, such as sacs and membranes, by self-assembly.22–24 In the herein presented work, we have investigated, for the first time, the interactions between these two molecules by QCM-D. This technique allows for marker-free measurement of specific interactions between immobilized molecules and analytes in solution and has been widely used for studying macromolecular interactions. Due to its sensitivity, it allows detecting mass changes of the order of ng cm−2, and permits us to measure the viscoelastic properties of the resulting film.36,37Fig. 2 shows the normalized frequency (Δfν/ν, where ν is the overtone) and dissipation (ΔD) variations for the 7th overtone (35 MHz). The first 15 minutes in Fig. 2 show the establishment of the baseline with 0.15 M NaCl. The subsequent 45 minutes correspond to the deposition of K3-PA, where a decrease in the frequency (of about 23 Hz) and an increase in ΔD were observed. After rinsing with NaCl, the hyaluronan solution was pumped continuously for 45 minutes, when an adsorption plateau was attained. A decrease in frequency of 38 Hz was registered, and again an increase in dissipation has occurred. After HA deposition, the film was again rinsed with NaCl. During this washing step, the variation in both Δf and ΔD was very small, indicating the formation of a stable film and a strong interaction between both molecules. The frequency variation, Δf, decreases with time, due to the deposition of peptide or hyaluronan on the quartz crystal. On the other hand, the dissipation, ΔD, increases, revealing that the film is not rigid and begins dissipating energy, thus exhibiting the typical viscoelastic behavior. This behavior has been observed before when studying the interaction between poly(L-lysine) and hyaluronan by QCM-D.38 The QCM-D results demonstrated and confirmed the strong interaction between the K3-PA and HA and the formation of a stable complex.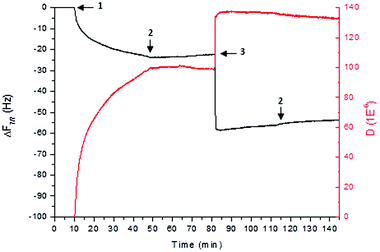 | ||
| Fig. 2 PA–HA interaction. QCM-D monitoring of frequency (Δf, black) and dissipation (ΔD, red) changes obtained at the 7th overtone, during deposition of peptide (step 1) and hyaluronan (step 3) on a bare crystal (step 2 relates to rising). The frequency of this overtone was normalized to the fundamental resonant frequency of the quartz crystal, by dividing it by ν (where ν = 7). | ||
Membrane microstructure and degradation
In this work, we aimed to recreate some aspects of the physical and biochemical environment of skin tissue. It was shown previously that hierarchical membranes can be formed by instant self-assembly between a positively charged PA (K3-PA) and the megadalton hyaluronan (HA) under certain conditions.22,23 We thought that these fibrillar membranes, containing components of skin ECM, could provide a starting platform to design complex biomimetic environments of skin ECM (Fig. 3). SEM analysis of the microstructure of the membranes revealed a very highly organized structure exhibiting two distinct surfaces, as observed previously.22,23,32 The surface corresponding to the HA side (Fig. 3C1) is characterized by a rough and amorphous structure, whereas the peptide side (Fig. 3C2) exhibits a network of organized nanostructures (nanofibers) randomly distributed, resembling the fibrillar structure of natural ECM. SEM micrographs of the cross section revealed a membrane with around 14 μm thickness and aligned nanofiber bundles perpendicular to the interface (Fig. 3C3). To investigate if the addition of the K3RGDS-PA would affect the membrane microstructure and morphology, SEM was used to image membranes containing 50% K3RGDS-PA. Membranes containing 50% K3RGDS-PA presented a similar thickness and microstructure to the ones formed with 100% K3-PA (Fig. S5, ESI†). Fluorescent micrographs obtained by confocal microscopy, using the fluorescently labeled K3KrhodRGDS-PA (red) and hyaluronan (green), showed a perfect overlap of the two, in yellow, resulting from the interaction between the peptide and hyaluronan (Fig. 3B2). This strong yellow region is also seen in a software simulated cross section (Fig. 3B1), surrounded by a less intense area in red or green, that corresponds to the peptide and hyaluronan sides, respectively. The uniform distribution of the red signal showed that RGDS sequence was well distributed within the membranes although the spatio control of the signal on specific locations of the membrane is still a major challenge.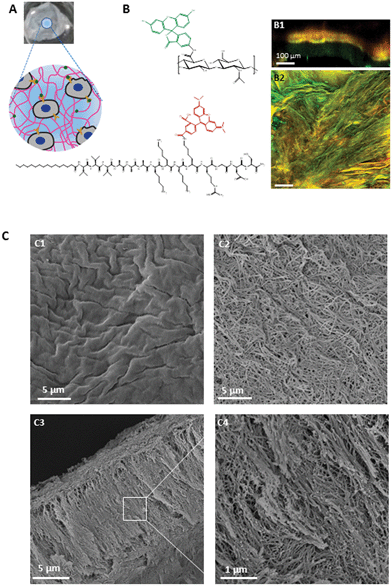 | ||
| Fig. 3 PA–HA membrane microstructure. (A) Schematic representation of PA–HA membranes functionalized with bioactive molecules (green) interacting with cell integrins (yellow). (B) Confocal microscopy images of the membranes prepared with 1% (w/v) fluorescein–HA and a 2% (w/v) peptide mixture containing 0.1% K3K(Rhod)RGDS-PA. Images show the localization of HA (green) and PA (red) over the membrane surface (B2) and cross section (B1). Yellow represents the overlaping of both components. (C) SEM micrographs of self-assembled membranes with 1% (w/v) HA and 2% (w/v) K3-PA, showing the surface on the polymer (C1) and peptide (C2) sides and cross section (C3, C4). | ||
The natural ECM is an extremely dynamic network that consists of protein fibers and glycosaminoglycans that support cell fate and provide biophysical and biochemical cues to cells through cell surface receptors, such as integrins.1,2,39 Cells degrade the ECM through proteases during their migration. Synthetic ECM matrices that aim to provide an environment for tissue regeneration should recapitulate key features of the natural ECM such as integrin mediated cell binding, cell migration and proliferation, while also allowing their degradation, offering a platform on which cell-triggered remodeling could occur. To mimic the turnover of natural ECM, our matrices were designed to be sensitive to enzymes expressed by surrounding cells (e.g. hyaluronidase). This cell-mediated degradation is a process reminiscent of tissue remodeling. It is well known that the degradation of hyaluronan into large oligosaccharides is mediated by hyaluronidase.40 Therefore, the degradation behavior of the PA–HA membranes was studied in three different conditions, PBS and PBS containing 2.6 U mL−1 and 50 U mL−1 HAase. Degradation was followed by the quantification of N-acetylglucosamine in solution and by SEM analysis (Fig. 4). Incubation in PBS up to 14 days did not cause degradation of the membranes since the amount of N-acetylamino sugars in the supernatant was significantly lower than that obtained for the membranes incubated in the higher enzyme concentration (50 U mL−1). Compared with the physiological concentration (2.6 U mL−1), only for the latest time point a statistical difference was observed. In addition, no evident signs of degradation are observed in the SEM images for the membranes incubated in PBS only. This result indicates that the membranes are relatively stable in buffer saline solutions, but are susceptible to enzymatic degradation by HAase. As expected, the membrane degradation was significantly enhanced when incubated with higher enzyme concentration (50 U mL−1) than in the presence of the enzyme under physiological conditions (2.6 U mL−1), as demonstrated by the higher amount of released N-acetylamino sugars (Fig. 4A). SEM analysis of the membranes collected at different time points corroborates these results, showing the appearance of pores on the membrane surface (Fig. 4B1) and cross-section (Fig. 4B2), as a result of HA degradation by HAase. At a higher HAase concentration (50 U mL−1), the degradation of the membranes is more evident, but progressive over time (Fig. 4B1–B2).
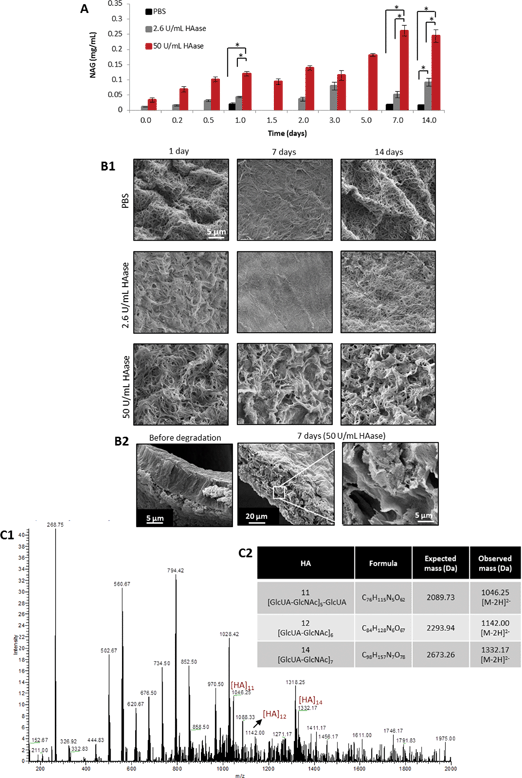 | ||
| Fig. 4 Degradation of PA–HA membranes. (A) Quantification of N-acetylamino sugars released from K3-HA membranes in PBS and PBS containing 2.6 U mL−1 and 50 U mL−1 HAase (*p < 0.05, error bars represent standard deviation (n = 3)). (B1) SEM images showing differences in membrane microstructure when exposed to different hyaluronidase (HAase) concentrations up to 14 days. (B2) Cross section of the membranes before and after exposure to 50 U mL−1 HAase evidencing that degradation is occurring not only on the surface but also inside the membrane. (C1) Negative ESI-MS of the supernatant after incubating the membranes in 2.6 U mL−1 HAase for 7 days showing the presence of HA fragments. (C2) Observed and theoretical molecular masses of HA oligosaccharides. | ||
The presence of HA oligosaccharides as degradation products was confirmed by mass spectroscopy. ESI-MS analysis of the supernatants after membrane degradation in a 2.6 U mL−1 HAase solution for 7 days showed three main peaks, corresponding to [HA]11: [M–2H]2−m/z = 1046.25, [HA]12: [M–2H]2−m/z = 1142 and [HA]14 [M–2H]2−m/z = 1332.17 (Fig. 4C).41 The observed mass for [HA]11 is a fragmentation product of ESI-MS since digestion with HAase produces even-numbered oligosaccharides. To confirm that the observed masses are not caused by the technique itself ESI-MS analysis of a HA solution was performed (Fig. S6, ESI†) but no known masses of HA oligosaccharides were identified.
The membranes herein presented were shown to degrade gradually over time in the presence of HAase at physiological concentration. In addition, it is expected that the peptide component of the membrane will degrade over time by hydrolytic and enzymatic processes into amino acids, which are nontoxic and easily cleared in the body. Peptides can be designed to be susceptible to enzymatic cleavage by incorporating matrix metalloproteases (MMPs)-sensitive peptide sequences,42,43 a strategy that is currently being explored in our lab. By having membranes sensitive to enzymatic activities, we are pursuing the goal of biomimetic matrix degradation. These PA–HA membranes may present an advantage over other systems, once slow degradation will enable the migration of the adhered cells, as well as matrix remodeling and new tissue formation led by the artificial ECM template.
Fibroblast response to the PA–HA self-assembled membranes displaying different densities of the RGDS epitope
Preliminary studies were performed with hDFb cultured on K3PA–HA membranes in DMEM with 10% FBS up to 7 days. These studies indicated that these membranes supported the adherence of fibroblasts, without presenting cytotoxicity (Fig. S7, ESI†).Understanding the factors that regulate cell behavior is important in many therapeutic applications. In most studies, cell behavior is studied in a culture medium supplemented with serum, which is poorly defined in terms of protein content. To investigate whether the produced PA–HA membranes could integrate cell-adhesive ligands and anchor human dermal fibroblasts, serum-free culture conditions were used to eliminate the complexity of the competing adsorption process of serum proteins on the membrane surface.
It has been shown that fibroblast survival, proliferation and migration are dependent on cell adhesion and that fibronectin (FN) binding domains have significant implications for skin wound healing.4 These activities require fibroblast attachment to the RGD sequence in the tenth FN type III repeat, via cell surface membrane integrin receptors.44 Several authors reported that inert biomaterials benefit from the presence of the RGDS signaling molecule for optimal cell recognition and adhesion.14–16,31 To enhance the adhesion of cells on the developed PA–HA membranes, the RGDS motif (which is a known ligand for integrin receptors) was incorporated into the PA structure creating adhesion points to support integrin-mediated cellular adhesion and migration on the membranes. Previous studies have shown that the density of the RGDS epitope impacts cell recognition and adhesion.45 Considering this, experiments were performed varying the K3RGDS-PA composition on the membranes (1, 10 and 50%) to determine the optimal RGDS density. Membranes containing 10% K3DGSR-PA or only K3-PA were used as controls.
DNA quantification results (Fig. 5A) showed that when seeded on 50% K3RGDS-PA containing membranes, hDFb adhered at a significantly higher number than the ones with a lower concentration of RGDS, 1 and 10%, or without RGDS, K3-PA and K3DGSR-PA for all time points except at 12 h of culture.
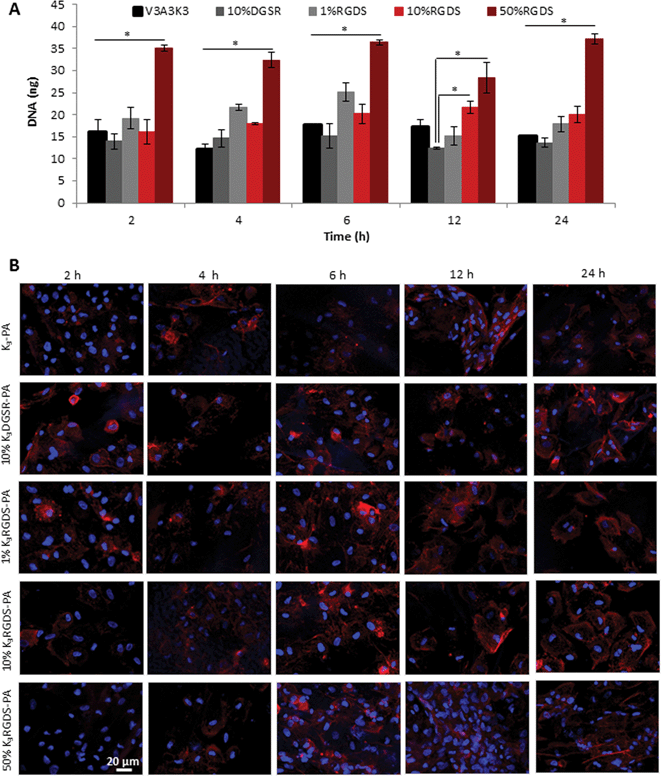 | ||
| Fig. 5 Cell adhesion on PA–HA membranes. dsDNA quantification (A) (*p < 0.05, error bars represent standard deviation (n = 3)) and confocal fluorescence images (B) of hDFb cultured on HA–PA membranes containing 1, 10 and 50% K3RGDS-PA or 10% (w/v) K3DGSR-PA up to 24 hours of culture. F-actin was labeled with TRITC-phalloidin (red) and nuclei with DAPI (blue). | ||
Cell morphology and distribution on the membranes was further examined by confocal microscopy analysis (Fig. 5B). After the first two hours, fibroblasts were uniformly distributed on the membrane surface and a large number of cells were observed on the 50% K3RGDS-PA containing membranes, as depicted by the higher density of nuclei. These results demonstrate the positive effect of the RGDS epitope on promoting cell adhesion, which is mediated by integrins on the cell surface. Considering the presence of HA in the membranes and knowing that CD44, a transmembrane receptor observed in a number of different cell types,46 mediates HA dependent cell adhesion, the expression of this receptor by hDFbs was assessed (ESI, Fig. S8†). Although these cells showed a high expression of CD44 (98.67%), cells cultured on HA-based hydrogels (ESI, Fig. S9†) did not show strong adhesion on these substrates. This result suggests that integrins may play a major role, rather than CD44, on cell adhesion on PA–HA membranes.
Staining of cell cytoskeleton (Fig. 5B) did not reveal strong organized actin fibers. Most of what is known about the cell structure and function in vitro derives from cells plated on rigid substrates, such as plastic or glass, laminated with a thin film of serum proteins. As a result, some prominent aspects of cell structure, such as the elongated morphology of cultured fibroblasts or the prevalence of large actin fibers that are commonly studied in vitro, are rarely if ever seen in vivo.47,48 Furthermore, cells cultured on soft substrates show diffuse adhesion complexes with poor actin cytoskeleton organization49 and the PA–HA membranes used in this study were shown to have low stiffness (0.9 MPa in the wet state).23
Previous findings have also shown that fibronectin fragments containing the RGD cell-binding domain alone cannot sustain cytoskeletal organization of human dermal fibroblasts,50 which might explain our observations.
Fibroblasts morphology observed in the SEM micrographs is similar to what has been observed previously by others on RGD-coated glass substrates.45 After 24 hours of culture, only the conditions with 10% and 50% K3RGDS-PA showed the majority of the cells fully adherent (Fig. 6). Under these conditions, fibroblasts had spread out exhibiting extended lamellipodia and filopodia interacting with fibrillar structure of the membrane surface, thus confirming strong adhesion to the substrate (Fig. 6a and b). In contrast, cells cultured on the surface of smooth HA hydrogel (ESI, Fig. S9†) were completely round, without showing cell protrusions (lamellipodia and filopodia) and did not flatten upon contact with the hydrogel surface.
 | ||
| Fig. 6 Cell morphology on PA–HA membranes. SEM micrographs of hDFb cultured on HA–PA membranes containing 1, 10 and 50% K3RGDS-PA or 10% (w/v) K3DGSR-PA. Cells were cultured on the PA side in medium without FBS up to 24 h. (a) and (b) show a higher magnification of cells on the surface of HA–PA membranes with 10 and 50% (w/v) RGDS, respectively. Black and white arrows indicate the presence of filopodia and lamellipodia, respectively. | ||
This result highlights the role of the nanofibrillar structure of the self-assembled membranes in controlling cell–matrix interactions. This is in accordance with what was reported in the literature that fibroblasts in 2D cultures normally exhibit a flat morphology with dorsal–ventral polarity and large lamellipodia.51 We should highlight that the cell culture experiments were performed in the absence of serum for the entire study and the membranes are displaying the RGDS cell-adhesive domain alone.
Further experiments are yet needed to investigate cell response to HA–PA membranes with higher stiffness and/or with other fibronectin fragment domains in order to be able to make additional considerations regarding the effect of each one of these variables on cell morphology.
4. Conclusion
This study has demonstrated that the 2D membranes, formed by self-assembly between hyaluronan and a positively charged peptide amphiphile, are susceptible to enzymatic degradation by hyaluronidase. In the presence of hyaluronidase at physiological concentration, the membranes slowly degrade over time. The gradual degradation of the membranes is important for allowing the migration of cells and/or the controlled release of bioactive molecules incorporated into the membrane that can control the function of attached cells. Membranes presenting the cell adhesive ligand RGDS showed to increase efficiently the attachment of fibroblasts. This capability to incorporate biochemical signals into 2D self-assembled membranes enables the study of cellular responses to physiologically relevant signal variations. Furthermore, these bioactive matrices permit cell culture studies without serum for short periods. We expect that the proposed biodegradable hybrid membranes could offer significant potential as a biomimetic bioactive supportive matrix for wound healing.Acknowledgements
Funding for this study was provided by the Portuguese Foundation for Science and Technology (FCT, grant PTDC/EBB-BIO/114523/2009). D. S. Ferreira gratefully acknowledges FCT for a PhD scholarship (SFRH/BD/44977/2008). We also thank Dr A. M. Azevedo from Instituto Superior Técnico (BERG, Lisbon, Portugal) for her support in obtaining circular dichroism data, and M. T. Cerqueira and I. M. Martins from the 3B's Research Group at the University of Minho (Portugal) for assistance in the isolation of human dermal fibroblasts and FACS analysis, respectively.References
- H. K. Kleinman, D. Philp and M. P. Hoffman, Role of the extracellular matrix in morphogenesis, Curr. Opin. Biotechnol., 2003, 14, 526–532, DOI:10.1016/j.copbio.2003.08.002.
- W. P. Daley, S. B. Peters and M. Larsen, Extracellular matrix dynamics in development and regenerative medicine, J. Cell Sci., 2008, 121, 255–264, DOI:10.1242/jcs.006064.
- F. G. Giancotti and E. Ruoslahti, Integrin signaling, Science, 1999, 285, 1028–1033, DOI:10.1126/science.285.5430.1028.
- M. S. Agren and M. Werthen, The extracellular matrix in wound healing: a closer look at therapeutics for chronic wounds, Int. J. Low. Extrem. Wounds, 2007, 6, 82–97, DOI:10.1177/1534734607301394.
- S. Sakai, R. Yasuda, T. Sayo, O. Ishikawa and S. Inoue, Hyaluronan exists in the normal stratum corneum, J. Invest. Dermatol., 2000, 114, 1184–1187 CrossRef CAS.
- D. Jiang, J. Liang and P. W. Noble, Hyaluronan as an immune regulator in human diseases, Physiol. Rev., 2011, 91, 221–264, DOI:10.1152/physrev.00052.2009.
- R. Stern and H. I. Maibach, Hyaluronan in skin: aspects of aging and its pharmacologic modulation, Clin. Dermatol., 2008, 26, 106–122, DOI:10.1016/j.clindermatol.2007.09.013.
- J. H. Collier, J. S. Rudra, J. Z. Gasiorowski and J. P. Jung, Multi-component extracellular matrices based on peptide self-assembly, Chem. Soc. Rev., 2010, 39, 3413–3424, 10.1039/b914337h.
- E. Ruoslahti and M. D. Pierschbacher, Arg-Gly-Asp: a versatile cell recognition signal, Cell, 1986, 44, 517–518 CrossRef CAS.
- M. D. Pierschbacher and E. Ruoslahti, Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule, Nature, 1984, 309, 30–33 CrossRef CAS.
- Y. D. Park, N. Tirelli and J. A. Hubbell, Photopolymerized hyaluronic acid-based hydrogels and interpenetrating networks, Biomaterials, 2003, 24, 893–900, DOI:10.1016/s0142-9612(02)00420-9.
- S. Khetan, J. S. Katz and J. A. Burdick, Sequential crosslinking to control cellular spreading in 3-dimensional hydrogels, Soft Matter, 2009, 5, 1601–1606, 10.1039/b820385g.
- J. A. Burdick and G. D. Prestwich, Hyaluronic acid hydrogels for biomedical applications, Adv. Mater., 2011, 23, H41–H56, DOI:10.1002/adma.201003963.
- U. Hersel, C. Dahmen and H. Kessler, RGD modified polymers: biomaterials for stimulated cell adhesion and beyond, Biomaterials, 2003, 24, 4385–4415, DOI:10.1016/s0142-9612(03)00343-0.
- J. P. Jung, A. K. Nagaraj, E. K. Fox, J. S. Rudra, J. M. Devgun and J. H. Collier, Co-assembling peptides as defined matrices for endothelial cells, Biomaterials, 2009, 30, 2400–2410, DOI:10.1016/j.biomaterials.2009.01.033.
- L. Perlin, S. MacNeil and S. Rimmer, Production and performance of biomaterials containing RGD peptides, Soft Matter, 2008, 4, 2331–2349, 10.1039/b801646a.
- K. Shroff, E. L. Rexeisen, M. A. Arunagirinathan and E. Kokkoli, Fibronectin-mimetic peptide-amphiphile nanofiber gels support increased cell adhesion and promote ECM production, Soft Matter, 2010, 6, 5064–5072, 10.1039/c0sm00321b.
- X. Zhao, F. Pan, H. Xu, M. Yaseen, H. Shan, C. A. E. Hauser, S. Zhang and J. R. Lu, Molecular self-assembly and applications of designer peptide amphiphiles, Chem. Soc. Rev., 2010, 39, 3480–3498 RSC.
- H. Cui, M. J. Webber and S. I. Stupp, Self-assembly of peptide amphiphiles: from molecules to nanostructures to biomaterials, Pept. Sci., 2010, 94, 1–18, DOI:10.1002/bip.21328.
- D. W. P. M. Lowik and J. C. M. van Hest, Peptide based amphiphiles, Chem. Soc. Rev., 2004, 33, 234–245 RSC.
- S. G. Zhang, Fabrication of novel biomaterials through molecular self-assembly, Nat. Biotechnol., 2003, 21, 1171–1178, DOI:10.1038/nbt874.
- D. Carvajal, R. Bitton, J. R. Mantei, Y. S. Velichko, S. I. Stupp and K. R. Shull, Physical properties of hierarchically ordered self-assembled planar and spherical membranes, Soft Matter, 2010, 6, 1816–1823, 10.1039/b923903k.
- R. M. Capito, H. S. Azevedo, Y. S. Velichko, A. Mata and S. I. Stupp, Self-assembly of large and small molecules into hierarchically ordered sacs and membranes, Science, 2008, 319, 1812–1816, DOI:10.1126/science.1154586.
- L. W. Chow, R. Bitton, M. J. Webber, D. Carvajal, K. R. Shull, A. K. Sharma and S. I. Stupp, A bioactive self-assembled membrane to promote angiogenesis, Biomaterials, 2011, 32, 1574–1582 CrossRef CAS.
- J. Gajewiak, S. S. Cai, X. Z. Shu and G. D. Prestwich, Aminooxy pluronics: synthesis and preparation of glycosaminoglycan adducts, Biomacromolecules, 2006, 7, 1781–1789, DOI:10.1021/bm060111b.
- T. Takahashi, M. Ikegami-Kawai, R. Okuda and K. Suzuki, A fluorimetric Morgan–Elson assay method for hyaluronidase activity, Anal. Biochem., 2003, 322, 257–263, DOI:10.1016/j.ab.2003.08.005.
- G. A. Silva, C. Czeisler, K. L. Niece, E. Beniash, D. A. Harrington, J. A. Kessler and S. I. Stupp, Selective differentiation of neural progenitor cells by high-epitope density nanofibers, Science, 2004, 303, 1352–1355, DOI:10.1126/science.1093783.
- M. J. Webber, J. Tongers, C. J. Newcomb, K. T. Marquardt, J. Bauersachs, D. W. Losordo and S. I. Stupp, Supramolecular nanostructures that mimic VEGF as a strategy for ischemic tissue repair (vol 108, pg 13438, 2011), Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9220–9220, DOI:10.1073/pnas.1207994109.
- M. J. Webber, J. Tongers, M. A. Renault, J. G. Roncalli, D. W. Losordo and S. I. Stupp, Development of bioactive peptide amphiphiles for therapeutic cell delivery, Acta Biomater., 2010, 6, 3–11, DOI:10.1016/j.actbio.2009.07.031.
- R. N. Shah, N. A. Shah, M. M. D. Lim, C. Hsieh, G. Nuber and S. I. Stupp, Supramolecular design of self-assembling nanofibers for cartilage regeneration, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3293–3298, DOI:10.1073/pnas.0906501107.
- H. Storrie, M. O. Guler, S. N. Abu-Amara, T. Volberg, M. Rao, B. Geiger and S. I. Stupp, Supramolecular crafting of cell adhesion, Biomaterials, 2007, 28, 4608–4618, DOI:10.1016/j.biomaterials.2007.06.026.
- R. H. Zha, S. Sur and S. I. Stupp, Self-assembly of cytotoxic peptide amphiphiles into supramolecular membranes for cancer therapy, Adv. Healthcare Mater., 2013, 2, 126–133, DOI:10.1002/adhm.201200118.
- A. C. Mendes, K. H. Smith, E. Tejeda-Montes, E. Engel, R. L. Reis, H. S. Azevedo and A. Mata, Co-assembled and microfabricated bioactive membranes, Adv. Funct. Mater., 2013, 23, 430–438, DOI:10.1002/adfm.201201065.
- X. M. Wang, A. Horii and S. G. Zhang, Designer functionalized self-assembling peptide nanofiber scaffolds for growth, migration, and tubulogenesis of human umbilical vein endothelial cells, Soft Matter, 2008, 4, 2388–2395, 10.1039/b807155a.
- H. A. Behanna, J. Donners, A. C. Gordon and S. I. Stupp, Coassembly of amphiphiles with opposite peptide polarities into nanofibers, J. Am. Chem. Soc., 2005, 127, 1193–1200, DOI:10.1021/ja044863u.
- R. R. Costa, C. A. Custodio, A. M. Testero, F. J. Arias, J. C. Rodriguez-Cabello, N. M. Alves and J. F. Mano, Stimuli-responsive thin coatings using elastin-like polymers for biomedical applications, Adv. Funct. Mater., 2009, 19, 3210–3218, DOI:10.1002/adfm.200900568.
- L. Y. Shen, P. Chaudouet, J. A. Ji and C. Picart, pH-amplified multilayer films based on hyaluronan: influence of HA molecular weight and concentration on film growth and stability, Biomacromolecules, 2011, 12, 1322–1331, DOI:10.1021/bm200070k.
- C. Picart, P. Lavalle, P. Hubert, F. J. G. Cuisinier, G. Decher, P. Schaaf and J. C. Voegel, Buildup mechanism for poly(L-lysine)/hyaluronic acid films onto a solid surface, Langmuir, 2001, 17, 7414–7424, DOI:10.1021/la010848g.
- J. A. Hubbell, Materials as morphogenetic guides in tissue engineering, Curr. Opin. Biotechnol., 2003, 14, 551–558, DOI:10.1016/j.copbio.2003.09.004.
- B. Smedsrod, Cellular events in the uptake and degradation of hyaluronan, Adv. Drug Delivery Rev., 1991, 7, 265–278 CrossRef.
- D. J. Mahoney, R. T. Aplin, A. Calabro, V. C. Hascall and A. J. Day, Novel methods for the preparation and characterization of hyaluronan oligosaccharides of defined length, Glycobiology, 2001, 11, 1025–1033 CrossRef CAS.
- K. M. Galler, L. Aulisa, K. R. Regan, R. N. D'Souza and J. D. Hartgerink, Self-assembling multidomain peptide hydrogels: designed susceptibility to enzymatic cleavage allows enhanced cell migration and spreading, J. Am. Chem. Soc., 2010, 132, 3217–3223, DOI:10.1021/ja910481t.
- J. Patterson and J. A. Hubbell, Enhanced proteolytic degradation of molecularly engineered PEG hydrogels in response to MMP-1 and MMP-2, Biomaterials, 2010, 31, 7836–7845, DOI:10.1016/j.biomaterials.2010.06.061.
- F. Lin, X.-D. Ren, Z. Pan, L. Macri, W.-X. Zong, M. G. Tonnesen, M. Rafailovich, D. Bar-Sagi and R. A. F. Clark, Fibronectin growth factor-binding domains are required for fibroblast survival, J. Invest. Dermatol., 2011, 131, 84–98 CrossRef CAS.
- S. P. Massia and J. A. Hubbell, An RGD spacing of 440 nm is sufficient for integrin alpha V beta 3-mediated fibroblast spreading and 140 nm for focal contact and stress fiber formation, J. Cell Biol., 1991, 114, 1089–1100, DOI:10.1083/jcb.114.5.1089.
- A. Aruffo, I. Stamenkovic, M. Melnick, C. B. Underhill and B. Seed, CD44 is the principal cell surface receptor for hyaluronate, Cell, 1990, 61, 1303–1313, DOI:10.1016/0092-8674(90)90694-A.
- T. Yeung, P. C. Georges, L. A. Flanagan, B. Marg, M. Ortiz, M. Funaki, N. Zahir, W. Y. Ming, V. Weaver and P. A. Janmey, Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion, Cell Motil. Cytoskeleton, 2005, 60, 24–34, DOI:10.1002/cm.20041.
- D. A. Lauffenburger and A. F. Horwitz, Cell migration: a physically integrated molecular process, Cell, 1996, 84, 359–369 CrossRef CAS.
- D. E. Discher, P. Janmey and Y.-l. Wang, Tissue cells feel and respond to the stiffness of their substrate, Science, 2005, 310, 1139–1143, DOI:10.1126/science.1116995.
- R. A. F. Clark, J.-Q. An, D. Greiling, A. Khan and J. E. Schwarzbauer, Fibroblast migration on fibronectin requires three distinct functional domains, J. Invest. Dermatol., 2003, 121, 695–705 CrossRef CAS.
- K. M. Yamada and E. Cukierman, Modeling tissue morphogenesis and cancer in 3D, Cell, 2007, 130, 601–610, DOI:10.1016/j.cell.2007.08.006.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3bm60019j |
| This journal is © The Royal Society of Chemistry 2013 |