Micro-patterning of streptavidin–biotin-ampicillin/heparin on poly(tetrafluoroethylene) (PTFE) surfaces: simultaneous antimicrobial and anticoagulant activity†
Nattharika
Aumsuwan
,
Heather A.
Pearson
and
Marek W.
Urban
*
Department of Materials Science and Engineering, Clemson University, Clemson, SC 29634, USA. E-mail: mareku@clemson.edu
First published on 2nd April 2013
Abstract
Formation of heterogeneous and controllable surface patterns on polymeric materials containing antimicrobial and anticoagulant components represent an attractive way of maintaining synthetic materials “clean” from adverse bio-activities. The primary surface “contaminants” are microbial films as well as blood coagulation, both affecting not only performance of internal or external devices, but often exhibiting detrimental effects on patients. In an effort to simultaneously inhibit formation of microbial films and surface blood coagulation multifunctional assemblies containing streptavidin (STR)–biotin bioconjugates were developed on poly(tetrafluoroethylene) (PTFE) surfaces. Using STR conjugated to a biotin-functionalized PTFE surface, spatially controlled micro-patterning was produced by grafting biotinylated polyethylene glycol (B-PEG) to COOH modified PTFE (MA-PTFE), followed by inkjet micro-printing of biotinylated ampicillin (B-AM) and biotinylated heparin (B-HP) molecules. These surfaces exhibit simultaneous antimicrobial and anticoagulant attributes manifested by “zone of inhibition” and anticoagulant measurements. Quantitative spectroscopic analysis revealed that the required surface density of COOH groups on PTFE is 2.94 × 10−7 g cm−2, and B-PEG and STR densities of 9.2 × 10−8 g cm−2 and 3.5 × 10−8 g cm−2, respectively, are sufficient to achieve simultaneous antimicrobial and anticoagulant responses. These studies also showed that the force required to remove STR–biotin conjugates attached to PTFE surfaces measured by atomic force microscopy is approximately 1090 pN, thus providing desirable surface mechanical stability.
Introduction
The ability to attach bioactive molecules to synthetic polymer surfaces enhancing biocompatibility provides a number of opportunities for the development of biosensor technologies,1–3 implants,4,5 tissue engineering,4,6–8 and other biomedical applications. Numerous methods have been introduced to enhance cell adhesion,9 alter blood compatibility,4,9 and anti-microbial10–13 or anti-fouling properties2,4,14,15 of polymeric surfaces. In this context, many naturally occurring proteins and polysaccharides16 have been utilized to obtain self-assembled mono-layers (SAMs),17 several synthetic approaches have utilized layer-by-layer deposition (lbl),11 photo-grafting,10,18 or surface microwave plasma reactions.13,19 Regardless of the approach, the primary objectives of many studies were to alter inert polymeric surfaces by attaching either biologically active species, or creating surfaces that would not adversely affect biological functions in contact with synthetic polymers.Among notable forces governing surface modifications, non-covalent attachments driven primarily by H-bonding, ionic interactions, van der Waals forces, or dipole–dipole interactions, dominated previous studies, but unfortunately, lifetime and stability of these relatively weak layers have found limited success. Ideally, one would like to enhance surface bonding and selectively attach surface species to usually non-reactive polymer surfaces in order to create a layer with simultaneous antimicrobial or anticoagulant responses. Ultimately, these properties have been recognized as necessary to halt often detrimental formation of bacterial colonies and blood clotting.
In view of these considerations, a well-known and unique interaction is the formation of streptavidin (STR)–biotin conjugates20,21via one of the strongest known non-covalent ligand–receptor forces22 manifested by bonding of four receptor sites with Kd values of 10−15 M.20,22 Due to inherent selectivity and the strength of this non-covalent interaction comparable to the strength of covalent bonding, combining this unique bonding with other covalently bonded surface components may lead to dual antibacterial and anticoagulant functions on inert polymeric surfaces. Taking advantage of the previous studies that have shown covalent attachments of multilayers (CAM) of penicillin,23 ampicillin,13 and heparin24 alternating with alternating PEG layers, these studies explore a novel approach of the step-wise attachment of the spatially resolved ampicillin and heparin molecules onto a PTFE surface via STR–biotin conjugates. In an effort to achieve lateral and vertical control of surface distribution of these components inkjet micro-patterning of biotinylated ampicillin (B-AM) and biotinylated heparin (B-HP) onto STR functionalized PTFE surfaces and biotinylated PEG (B-PEG) will be utilized.
Experimental section
Surface biotinylation and streptavidin immobilization
Medical grade PTFE specimens (McMaster-Carr, Atlanta, GA) were cleaned in an ultrasonic washer with a mixture of acetone and isopropanol for 30 min, dried, and kept in the desiccator. Plasma reactions were conducted using open reactor conditions, as described elsewhere. The PTFE substrate and powdered maleic anhydride (MA) (Aldrich Chemical Co.) were placed into the microwave reactor chamber and spaced 8.5 cm apart of each other. In a typical experiment, the reactor was evacuated to 150 mTorr, followed by purging it with Ar gas to reach a steady-state pressure of 250 mTorr at a flow rate of 3 mL min−1. At this point, microwave radiation at 600 W of power with an output frequency of 2.45 GHz was applied to the reactor to induce plasma formation for 7 seconds. Under these conditions, the reaction chamber pressure increased continuously during plasma discharge. After the reactions, specimens were washed in boiling water for 30 min to ensure that the newly formed species were not physisorbed on the surface and all maleic anhydride (MA) surface groups were converted to COOH groups. The carboxylic acid primed PTFE surfaces (MA-PTFE) were stored in a desiccator at ambient temperature.EZ-link® amine-PEG-biotin (B-PEG) (Thermo Scientific, Rockford, IL) was attached to the MA-PTFE surfaces by amine reactions with surface carboxylic acid groups. The PTFE was placed in 0.1 M 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) (Sigma Aldrich) for 4 h (pH 4–6), then the EDC activated surface was reacted with 1 mM B-PEG for 18 h. Such B-PEG–MA-PTFE specimens were removed, and washed with deionized water (DI) for 20 min and dried in a desiccator. STR was immobilized on the surfaces by incubation of the B-PEG–MA-PTFE specimens overnight in 30 μg mL−1 STR (Thermo Scientific, Rockford, IL) in phosphate-buffered saline (PBS) pH 7.4. The specimens with immobilized STR (STR–B-PEG–MA-PTFE) were subsequently rinsed in PBS and stored at 4 °C.
Ampicillin biotinylation and biotin–streptavidin recognition
B-AM was prepared by dissolving 0.06 g of ampicillin (AM) in 30 mL of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of dimethyl sulfoxide (DMSO) and water, then mixing the solution with excess 0.03 M N-hydroxysuccinimide ester of biotin (NHS-biotin) (Sigma Aldrich). The mixture was gently stirred for 6 h at room temperature. The reaction was terminated by adding Affi-Gel 102 (Bio-Rad Laboratories, Richmond, CA) in excess with stirring for 3 h. B-AM was separated from the solution by centrifugation and freeze dried under a vacuum for 2 days. The B-AM was always freshly prepared before use. The B-AM formation was confirmed by ATR FT-IR and NMR in Fig. S3 of the ESI.†
:1 mixture of dimethyl sulfoxide (DMSO) and water, then mixing the solution with excess 0.03 M N-hydroxysuccinimide ester of biotin (NHS-biotin) (Sigma Aldrich). The mixture was gently stirred for 6 h at room temperature. The reaction was terminated by adding Affi-Gel 102 (Bio-Rad Laboratories, Richmond, CA) in excess with stirring for 3 h. B-AM was separated from the solution by centrifugation and freeze dried under a vacuum for 2 days. The B-AM was always freshly prepared before use. The B-AM formation was confirmed by ATR FT-IR and NMR in Fig. S3 of the ESI.†
The patterned arrays of B-AM (synthesized) and B-HP (purchased from Sigma Aldrich) were generated with inkjet printing to create ∼10–20 μm diameter dots. 10 μg mL−1 of B-AM and B-HP aqueous solution were utilized as an ink for printing on the STR–B-PEG–MA-PTFE surfaces. After printing, the specimens set overnight, followed by rinsing with DI water, and drying in a desiccator.
Inkjet printing was conducted on a piezoelectric drop on demand (DOD) inkjet printer (Jetlab4) manufactured by Microfab Technologies Inc. (Plano, TX). The printing area was approximately 70 × 70 mm with an adjustable height (Z). The sample was held on the stage and polymer ink was printed through the 60 and 20 μm printhead orifice.
The modification of AFM tip was carried out by soaking the Si3N4 cantilever (Veeco Probes, CA) with 1 μg mL−1 of biotinylated bovine serum albumin (B-BSA) solution (Thermo Scientific, Rockford, IL) overnight at 37 °C. The cantilever was rinsed with PBS solution and dried on a glass slide.25
A scanning electron microscope (SEM) Quanta FEI series 200 FEG was used to evaluate the AFM tip before and after B-BSA immersion. All specimens were sputter coated with gold and analyzed at a 45° angle with a scanning electron beam.
Antimicrobial activity
To determine anti-microbial activity of multifunctional surfaces containing B-AM and B-HP (B-AM/B-HP–STR–B-PEG–MA-PTFE), each specimen was contacted with cultures of Staphylococcus aureus (S. aureus) (RN 6390). S. aureus bacteria was incubated for 4 hours at 37 °C in Triptic Soy Broth (TSB). The growth of bacteria was determined by the optical density (OD) data analysis, measured from the absorbance at 600 nm using a UV-vis spectrometer. A 50 μL of S. aureus culture was combined with 20 mL of fresh TSB and 0.7% of Triptic Soy Agar (TSA). This culture was then spread over individual TSA plates containing specimens from each reaction step to grow colonies. The typical size of each polymer specimen was 20 × 0.9 × 1.5 mm. PTFE, MA-PTFE, B-PEG–MA-PTFE, STR–B-PEG–MA-PTFE were used as controls. B-AM and B-HP solutions were printed in two distinct regions of the same specimen as well as using an alternating checkerboard patterning. Two additional controls printed only with B-AM or B-HP solutions were proceeded in order to determine the antimicrobial activity of the individual species. After incubating the TSA plates at 37 °C for 16 h, the antimicrobial activity of each specimen was determined.Anticoagulant activity
Lagomorph blood was collected by venipuncture and transferred into a syringe containing heparin (10 μL mL−1 for blood contacting experiments). The use of laboratory animals followed the NIH guidelines. Heparin attached samples (B-HP–STR–B-PEG–MA-PTFE), and controls were placed into 2.5 mL vials without any additive and filled with heparinized lagomorph blood followed by incubation at 37 °C for 2 h on a hematology mixer (Fisher Scientific, Pittsburgh, PA). Samples were rinsed with deionized water to remove any non-adhered blood from the surfaces. Optical images were immediately collected for each sample.Raman spectra were obtained using a Renishaw Raman microscope/spectrometer equipped with a computer controlled three-axis encoded motorized stage, a RenCam CCD detector, and a Leica microscope (DMLM series). The 785 nm diode laser provided an excitation source with a maximum power output of 300 mW. The samples, as well as, blood reference were placed on a gold slide and each Raman spectra was collected at 30 mW laser power at an acquisition time of 10 s. Raman imaging was carried out on each sample at 3 mW laser power for 1 min by tuning into the 1620 cm−1 band characteristic of the N–H vibrations of blood.
Surface characterization
Attenuated total reflectance Fourier transform infrared (ATR FT-IR) spectra were collected using a Bio-Rad FTS-6000 FT-IR single-beam spectrometer set at a 4 cm−1 resolution equipped with a deuterated triglycine sulphate (DTGS) detector and a 45° face angle Ge crystal. Each spectrum represents 400 co-added scans ratioed against a reference spectrum obtained by recording 400 co-added scans of an empty ATR cell. All spectra were corrected for spectral distortions using Q-ATR software. Knowing the extinction coefficient of the N–H vibrations, quantitative analysis was performed Urban–Huang algorithm. In this algorithm, the absorption index spectrum is refined by an iterative process that minimizes the difference between the true and calculated reflectivity resulting from optical effects, while maintaining the exact Kramers–Kronig relation between absorption (k) and refractive index (n).26,27 This iterative process was used in conjunction with the numerical Kramers–Kronig transformation (KKT) method to obtain ATR spectra suitable for quantitative analysis using the Beer–Lambert law. Further details involved in this algorithm have been published elsewhere.271H nuclear magnetic resonance (NMR) spectra were acquired using the Varian Mercury 300 MHz NMR spectrometer. Samples were prepared (5 mg mL−1) in chloroform (CDCl3), and spectra were recorded at room temperature. Typical acquisition parameters were a 45° pulse, 5 s relaxation delays, and 2 s collection with 256 repetitions.
Atomic force microscopy (AFM) measurements were analyzed on a Nanoscope IIIa Dimension 3000 scanning probe microscope, Digital Instruments. A silicon probe with 125 μm long silicon cantilever, nominal force constant of 60 N m−1 and resonance frequency of 200 kHz were used in a tapping mode, allowing estimation of surface topography and roughness.
Internal reflection IR imaging (IRIRI) experiments were conducted on a Varian Stingray system with a Ge internal reflection element allowing spatial resolution of about 1 μm. This system consists of a Varian FTS 7000 spectrometer, an UMA 600 FT-IR microscope with a focal plane array (FPA) image detector, and a semi-spherical Ge crystal. IRIR images were collected using the following spectral acquisition parameters: under sampling ratio of 2, rapid scan speed of 5 kHz, and 8 cm−1 spectral resolution. Image processing was performed using the Environment for Visualizing Images (ENVI) software (Research Systems, Inc., version 3.5). When appropriate, baseline correction algorithms were applied to compensate for baseline deviations accomplished by built-in application software supplied by GRAMS/AI v7.02 (Thermo Galactic).
Results and discussions
Using a simple inkjet printing technology with a lateral spatial resolution of ∼20 μm we reacted sequential layers of B-AM and B-HP onto a COOH-modified PTFE surface. In the first step of creating controllable micro-patterned multi-functional PTFE surfaces we reacted a surface anchor in the form of COOH groups, followed by a sequence of surface reactions of B-PEG, STR, B-AM and B-HP. Fig. 1a, steps A–E, illustrate a schematic diagram of steps involved in these experiments: A – microwave plasma reactions were utilized to covalently attach COOH groups onto a PTFE surface; B – B-PEG was reacted to COOH; C – STR was immobilized by biotin–STR conjugate formation; D – and E – B-HP (D) and B-AM (E) were attached to a biotin–STR conjugate. Fig. 1b illustrates ATR FT-IR spectra corresponding to each step shown in Fig. 1a. While Trace A illustrates the spectrum of the PTFE substrate, formation of COOH groups on the PTFE surfaces (MA-PTFE) is shown in Trace B, where the new band at 1720 cm−1 due to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (acid)28 is detected. The covalent attachment of B-PEG to the COOH–PTFE surfaces (B-PEG–MA-PTFE) with the aid of carbodiimide is manifested in Trace C by the presence of the band at 1650 cm−1 due to amide linkages26 resulting from the reactions of surface COOH with NH2 groups of B-PEG. The bands in Trace C at 1715 cm−1 due to the CO of biotin ureido ring26 and 1560 cm−1 due to amide N–H are responsible for the immobilization of B-PEG onto MA-PTFE surface. Also, the band at 1080 cm−1 due to C–O–C of PEG is detected in the 1300–1000 cm−1 region shown in Fig. S1 of the ESI.† The biotinylated surfaces were then immobilized in STR solutions resulting in the STR–biotin binding to form a STR–B-PEG–MA-PTFE surface complex. The bands at 1660, 1635 and 1535 cm−1 in Trace D due to the CO (amide I) and N–H (amide II) bands of the STR protein are detected, confirming the presence of STR on the surface. The last steps, D and E, result in the formation of B-HP and B-AM, respectively, on the STR–B-PEG–MA-PTFE surface which is confirmed by the presence of the 1630 cm−1 band due to OH bending of HP (Trace E) as well as the band at 1775 cm−1 due to CO (β-lactam) of B-AM (Trace F).
O (acid)28 is detected. The covalent attachment of B-PEG to the COOH–PTFE surfaces (B-PEG–MA-PTFE) with the aid of carbodiimide is manifested in Trace C by the presence of the band at 1650 cm−1 due to amide linkages26 resulting from the reactions of surface COOH with NH2 groups of B-PEG. The bands in Trace C at 1715 cm−1 due to the CO of biotin ureido ring26 and 1560 cm−1 due to amide N–H are responsible for the immobilization of B-PEG onto MA-PTFE surface. Also, the band at 1080 cm−1 due to C–O–C of PEG is detected in the 1300–1000 cm−1 region shown in Fig. S1 of the ESI.† The biotinylated surfaces were then immobilized in STR solutions resulting in the STR–biotin binding to form a STR–B-PEG–MA-PTFE surface complex. The bands at 1660, 1635 and 1535 cm−1 in Trace D due to the CO (amide I) and N–H (amide II) bands of the STR protein are detected, confirming the presence of STR on the surface. The last steps, D and E, result in the formation of B-HP and B-AM, respectively, on the STR–B-PEG–MA-PTFE surface which is confirmed by the presence of the 1630 cm−1 band due to OH bending of HP (Trace E) as well as the band at 1775 cm−1 due to CO (β-lactam) of B-AM (Trace F).
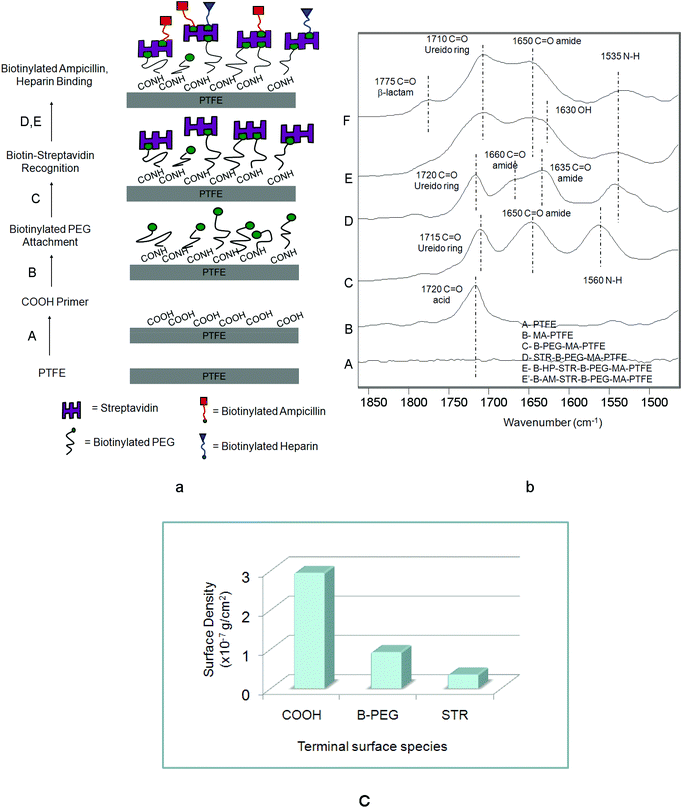 | ||
| Fig. 1 (a) Schematic diagram of surface reactions on PTFE: (A) microwave Ar plasma reactions leading to the formation of COOH groups, (B) attachment of B-BEG to COOH–PTFE surface, (C) immobilization of STR via STR–biotin conjugates, (D) B-HP binding to STR–B-PEG–MA-PTFE, (E) B-AM HP binding to STR–B-PEG–MA-PTFE; (b) ATR FT-IR spectra in the 1900–1400 cm−1 region: Trace A – PTFE, Trace B – MA-PTFE, Trace C – B-PEG–MA-PTFE, Trace D – STR–B-PEG–MA-PTFE, Trace E – B-HP–STR–B-PEG–MA-PTFE, Trace F – B-AM–STR–B-PEG–MA-PTFE; (c) surface density obtained from quantitative analysis of COOH, B-PEG, STR. | ||
In an effort to determine if these surface reactions result in stable, covalently bonded species, after each reaction step the specimen was boiled in water for 30 min to eliminate all physisorbed species. The spectroscopic analysis after boiling confirmed the covalent attachment after each step of the reaction and IRIR images (Fig. S2†) show that identical IR spectra were obtained before and after boiling. Also, to determine surface density, quantitative analysis performed after each step using Kramers–Kronig transformation (KKT) and the algorithm for quantitative ATR-FTIR spectroscopy24,27 for the bands at 1715, 1080, and 1640 cm−1 due to COOH, PEG, and STR were applied.20,23 The surface densities of COOH, B-PEG and STR layers were found to be 2.94 × 10−7 g cm−2, 9.2 × 10−8 g cm−2, and 3.5 × 10−8 g cm−2, respectively. The surface density values plotted as a function of each terminal group are shown in Fig. 1c.
While the above results show the covalent attachment of multilayers (CAM),29 another objective of these studies was to create patterned surfaces which exhibit laterally patterned morphologies. The motivation behind this approach is to generate not only surfaces with continually accessible antimicrobial and anticoagulant features effective against microbial film formation and inhibiting blood adhesion, but also to achieve horizontally controlled surface morphologies. For that reason B-AM and B-HP were inkjet printed onto the PTFE surface to obtain multi-patterned, multi-functional surfaces, such as illustrated in Fig. 2a. After printing, optical images shown in Fig. 2a′ were obtained, and to confirm the controllability of the process, B-AM and B-HP were printed onto STR–B-PEG–MA-PTFE surfaces and IRIR images were recorded. While Fig. 2a′′ illustrates the side-by-side B-AM and B-HP patterns, Fig. 2b, b′, c, and c′ show IRIR images as well as IR spectra recorded from the printed B-AM and B-HP surfaces. As seen in Fig. 2b′, upon tuning to the band characteristic of B-AM at 1780 cm−1, the spherical dark areas A, B, and C in Fig. 2b exhibit higher intensities compared to the areas D and E. Similarly, the spectra shown in Fig. 2c′ exhibit the highest intensities collected from the areas A′, B′, C′, and D′ (Fig. 2c), where the B-HP species are present, upon tuning into the band characteristic of B-HP at 1630 cm−1. These results confirm that B-AM and B-HP were printed in rows with a controllable horizontal side-by-side fashion.
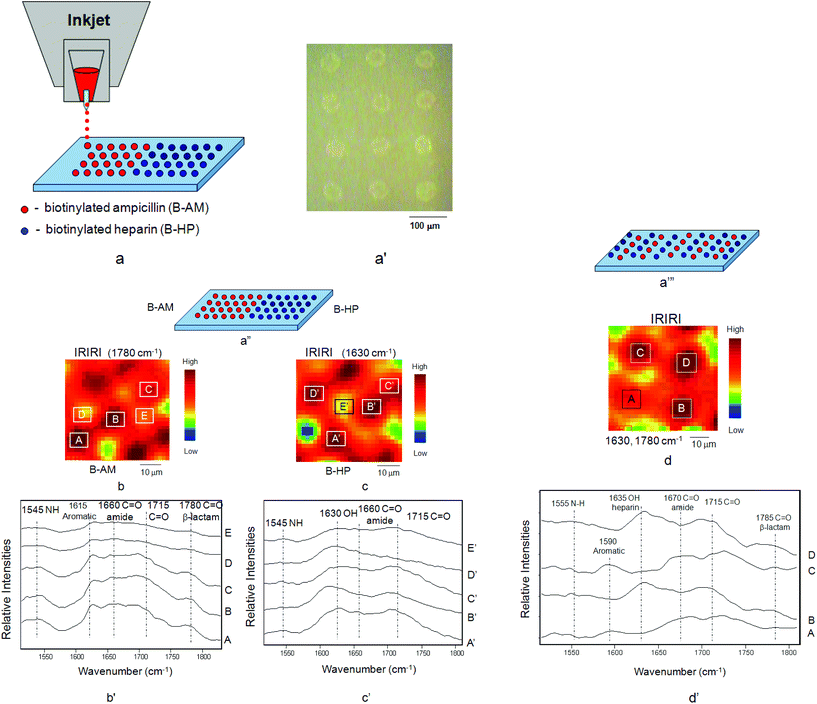 | ||
| Fig. 2 (a) Inkjet printing of B-AM and B-HP onto STR–B-PEG–MA-PTFE surface, (a′) optical images of B-AM and B-HP printed from the inkjet, (a′′) horizontal printing pattern of B-AM and B-HP, (b) IRIRI images of B-AM, (b′) IR spectra recorded from selected areas A, B, C, D, E of IRIR images of B-AM, (c) IRIRI images of B-HP, (c′) IR spectra recorded from selected areas A′, B′, C′, D′, E′ of IRIR images of B-HP, (a′′′) alternating stripe printing pattern of B-AM and B-HP, (d) IRIRI images of B-AM and B-HP, (d′) IR spectra recorded from selected areas A, C of IRIR images of B-AM and areas B, D of IRIR images of B-HP. | ||
An alternating stripe pattern, shown in Fig. 2a′′′, consisting of B-AM and B-HP dots was also printed. As seen in Fig. 2d, tuning to the band 1630 cm−1 of B-HP results in the spherical dark spots, with the areas labeled B and D showing enhanced intensities compared to the spectra collected from areas A and C. In contrast, the spectra collected from the areas A and C exhibit enhanced intensities of the bands at 1670 and 1590 cm−1 attributed to CO (amide) and C–C aromatic of B-AM, respectively. These results confirm again that B-AM and B-HP exhibit alternating printed patterns. It should be noted that the band at 1785 cm−1 was also detected in the spectra collected from the areas B and D where B-HP were printed, indicating the pattern overlap, or a likely hood of the mixing of B-AM and B-HP spots.
In an effort to determine forces necessary for detaching B-HP and B-AM from surfaces, AFM experiments were conducted in which dynamic recognition force mapping of the STR–B-PEG–MA-PTFE surface with a biotinylated AFM tip was analyzed. This is shown in Fig. 3. We immobilized biotinylated bovine serum albumin (B-BSA) on a silicon nitride AFM tip. The B-BSA protein was chosen in these model experiments in place of B-AM and B-HP due to its sufficiently stronger bond to the AFM tip.25Fig. 3a and b, illustrate SEM images of the AFM tip before and after immobilization of B-BSA. As seen in Fig. 3b, immobilization of B-BSA is manifested by the presence of the materials’ residue on the AFM tip. The recognition images obtained by simultaneous oscillating AFM tip modified with B-BSA on PTFE and STR–B-PEG–MA-PTFE surfaces are illustrated in Fig. 3b′ and c′. For unmodified PTFE, no significant features are observed. However, the STR–B-PEG–MA-PTFE recognition between biotin and STR is illustrated by the dark spots in the AFM image shown in Fig. 3c′ which are attributed to a decrease of the oscillation of the AFM tip caused by STR–biotin conjugates formation.
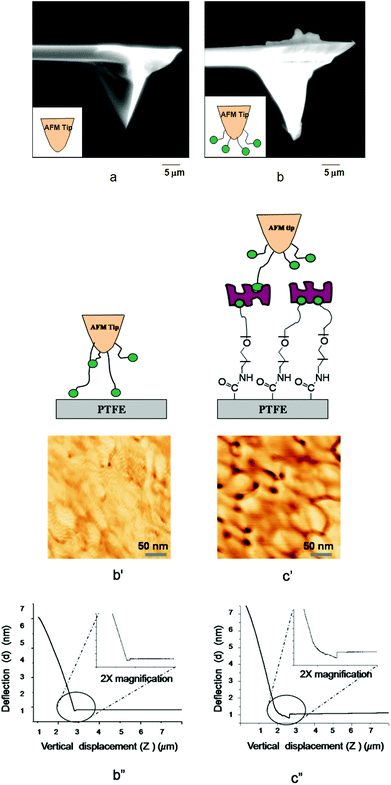 | ||
| Fig. 3 SEM images of (a) Si3N4·AFM tip before modification, (b) after modification with B-BSA; AFM dynamic force recognition of (b′) PTFE, (c′) STR–B-PEG–MA-PTFE; voltage displacement curve of (b′′) PTFE, (c′′) STR–B-PEG–MA-PTFE. | ||
While the results of these model experiments confirm that STR–B-PEG–MA-PTFE surfaces are capable of recognizing biotinylated B-AM and B-HP, the measurements of molecular recognition forces of STR–biotin obtained from AFM measurements will provide further information regarding surface adhesion forces. Fig. 3b′′ and c′′ illustrate the plot of cantilever deflection (d) as a function of the vertical displacement of the piezoelectric scanner (z). As seen in Fig. 3b′′, only minute deflection is observed for unmodified PTFE surfaces. In contrast, for STR–B-PEG–MA-PTFE surfaces the magnitude of deflection is ∼8 nm, resulting from the adhesion forces due to recognition of STR–biotin. This is illustrated in Fig. 3c′′. The cantilever deflection data obtained in these experiments converted into adhesion force (F) following the Hooke's law (F = kd, F is the adhesion force, k is the spring constant, d is the deflection, and the adhesion force of B-BSA and STR–B-PEG–MA-PTFE shown in Fig. 3c′′) results in a force of 1090 pN. Assuming that the unbinding force to rupture a single STR–biotin interactions is 80–100 pN,30,31 these experiments indicate that ∼11–13 pairs of STR–biotin conjugates are detached in one cantilever lift from the surface.
As stated earlier, an ultimate goal of these studies was to create simultaneous antimicrobial and anticoagulant responses on PTFE surfaces. The antimicrobial activity of B-AM and B-HP bound to the STR–B-PEG–MA-PTFE surfaces was evaluated by exposing respective specimens to S. aureus bacteria. Fig. 4a–h, illustrates optical images of TSA plates containing: (a) PTFE; (b) MA-PTFE; (c) B-PEG–MA-PTFE; (d) STR–B-PEG–MA-PTFE; (e) B-AM–STR–B-PEG–MA-PTFE; (f) B-HP–STR–B-PEG–MA-PTFE; (g) B-AM/B-HP–STR–B-PEG–MA-PTFE side-by-side pattern; and (h) B-AM/B-HP–STR–B-PEG–MA-PTFE alternating stripe pattern. As seen, (a) PTFE, (b) MA-PTFE, (c) B-PEG–MA-PTFE, and (d) STR–B-PEG–MA-PTFE do not exhibit antimicrobial activity. In contrast, when B-AM (Fig. 4e) and B-HP (Fig. 4f) were printed onto STR–B-PEG–MA-PTFE, a “zone of inhibition” is observed for B-AM (Fig. 4e), but not for B-HP (Fig. 4f). Similarly, for side-by-side printing of B-AM and B-HP (Fig. 4g), the B-AM containing side exhibits a “zone of inhibition”, while the B-HP printed side does not. When B-AM and B-HP were printed in an alternating stripe pattern (Fig. 4h), a “zone of inhibition” surrounding the specimen confirms the effectiveness of B-AM in killing S. aureus.
 | ||
| Fig. 4 Optical images of TSA plates containing (a) PTFE, (b) MA-PTFE, (c) B-PEG–MA-PTFE, (d) STR–B-PEG–MA-PTFE, (e) B-AM–STR–B-PEG–MA-PTFE, (f) B-HP–STR–B-PEG–MA-PTFE, (g) B-AM/B-HP–STR–B-PEG–MA-PTFE with side-by-side pattern, (h) B-AM/B-HP–STR–B-PEG–MA-PTFE with alternating stripe pattern after incubation with S. aureus for 16 h at 37 °C. | ||
Anticoagulant activity of B-HP bound to the STR–B-PEG–MA-PTFE surface was examined using Raman imaging tuned to 1620 cm−1. This band is due to N–H deformation vibrations attributed to hemoglobin protein constituents.32Fig. 5 shows Raman images of the specimens obtained from incubating each surface in lagomorph blood, in which PTFE (a), MA-PTFE (b), STR–B-PEG–MA-PTFE (c), and B-HP–STR–B-PEG–MA-PTFE (d) are Raman images of the substrates before blood exposure. While PTFE (a′), MA-PTFE (b′), and STR–B-PEG–MA-PTFE (c′), did not exhibit any anticoagulant activity, B-HP–STR–B-PEG–MA-PTFE (d′) did prevent clotting. For comparison, Fig. 5e shows the Raman image of dried lagomorph blood at 1620 cm−1. The effectiveness of HP attached to PTFE surfaces via biotin streptavidin interactions against blood coagulation is apparent. It should be noted that the choice of Raman imaging provided the opportunity for chemical analysis of minute quantities of all blood components, which are often not detected by scanning electron microscopy (SEM).32 Moreover, Raman spectra were collected on whole blood, which contains various clotting factors and is not limited to erythrocytes.
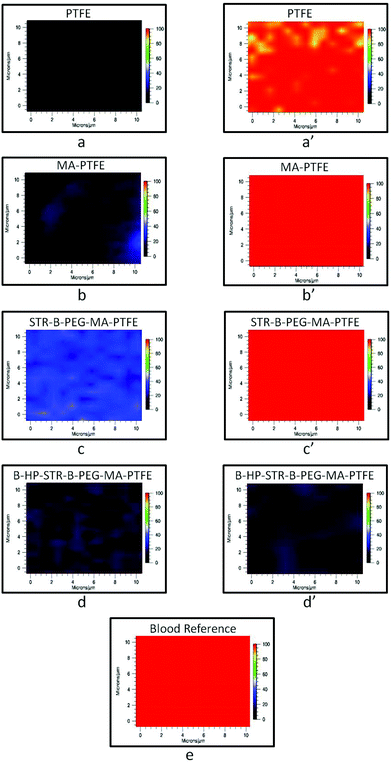 | ||
| Fig. 5 Raman images of (a) PTFE, (b) MA-PTFE, (c) STR–B-PEG–MA-PTFE, (d) B-HP–STR–B-PEG–MA-PTFE surfaces before anticoagulant testing and (a′) PTFE, (b′) MA-PTFE, (c′) STR–B-PEG–MA-PTFE, (d′) B-HP–STR–B-PEG–MA-PTFE surfaces after incubation in lagomorph blood for 2 h at 37 °C; Raman image of (e) lagomorph blood. | ||
Conclusions
Controllable micro-patterned multi-functional PTFE surfaces generated by the simultaneous inkjet printing of B-AM and B-HP, obtained by microwave plasma reactions generating COOH groups on the PTFE surface, followed by the reactions of B-PEG and STR immobilization, exhibit antimicrobial and anticoagulant activities as demonstrated for the B-AM printed areas that effectively kill S. aureus bacteria, while B-HP reacted areas that inhibit blood coagulation and adhesion. Concentration levels of surface layers manifested by surface density are 2.94 × 10−7 g cm−2 for COOH–PTFE, B-PEG and STR are 9.2 × 10−8 g cm−2 and 3.5 × 10−8 g cm−2, respectively, are sufficient to achieve antimicrobial and anticoagulant properties. Micro-patterned areas of ∼20 μm of B-AM and B-HP, using side-by-side and alternating stripe patterning, provides simultaneous access to antimicrobial and anticoagulant properties.Acknowledgements
This work was partially supported by NIH (UOICA15815) and by DMR (0215873) Major Instrumental Program. The authors thank S. Messer, B. Achord and C. Sahagun for technical assistance. J. Rich and N. Young of The University of Southern Mississippi Animal Resources are acknowledged for providing fresh lagomorph blood samples.References
- T. M. Squires, R. J. Messinger and S. R. Manalis, Nat. Biotechnol., 2008, 26, 417 CrossRef CAS.
- J. M. Goddard and J. H. Hotchkiss, Prog. Polym. Sci., 2007, 32, 698 CrossRef CAS.
- S. R. Lee, K. Sawada, H. Takao and M. Ishida, Biosens. Bioelectron., 2008, 24, 650 CrossRef CAS.
- T. Desmet, R. Morent, N. D. Geyter, C. Leys, E. Schacht and P. Dubruel, Biomacromolecules, 2009, 10, 2351 CrossRef CAS.
- W. T. S. Huck, Nat. Mater., 2005, 4, 271 CrossRef CAS.
- A. J. Putnam and D. J. Mooney, Nat. Med., 1996, 2, 824 CrossRef CAS.
- L. G. Griffith and G. Naughton, Science, 2002, 295, 1009 CrossRef CAS.
- H. Kim, J. Doh, D. J. Irvine, R. E. Cohen and P. T. Hammond, Biomacromolecules, 2004, 5, 822 CrossRef CAS.
- A. G. Kidane, H. Salacinski, A. Tiwari, K. R. Bruckdorfer and A. M. Seifalian, Biomacromolecules, 2004, 5, 798 CrossRef CAS.
- M. C. Lawson, R. Shoemaker, K. B. Hoth, C. N. Bowman and K. S. Anseth, Biomacromolecules, 2009, 10, 2221 CrossRef CAS.
- J. A. Lichter, K. J. V. Vliet and M. F. Rubner, Macromolecules, 2009, 42, 8573 CrossRef CAS.
- P. Kurt, L. Wood, D. E. Ohman and K. J. Wynne, Langmuir, 2007, 23, 4719 CrossRef CAS.
- N. Aumsuwan, M. McConnell and M. W. Urban, Biomacromolecules, 2009, 10, 623 CrossRef CAS.
- C. M. Yam, M. Deluge, D. Tang, A. Kumar and C. Cai, J. Colloid Interface Sci., 2006, 296, 118 CrossRef CAS.
- J. M. Harris, Introduction to Biotechnology and Biomedical Applications of Poly(ethylene glycol), Plenum Press, New York, 1992 Search PubMed.
- Z. G. Wang, L. S. Wan and Z. K. Xu, J. Membr. Sci., 2007, 304, 8 CrossRef CAS.
- N. P. Westcott and M. N. Yousaf, Langmuir, 2008, 24, 2261 CrossRef CAS.
- J. Deng, L. Wang, L. Liu and W. Yang, Prog. Polym. Sci., 2009, 34, 156 CrossRef CAS.
- W.-S. Bae and M. W. Urban, Langmuir, 2006, 22, 10277 CrossRef CAS.
- N. Malmstadt, D. E. Hyre, Z. Ding, A. S. Hoffman and P. S. Stayton, Bioconjugate Chem., 2003, 14, 575 CrossRef CAS.
- A. G. Young, A. J. McQuillan and D. P. Green, Langmuir, 2009, 25, 7416 CrossRef CAS.
- N. A. Lapin and Y. J. Chabal, J. Phys. Chem. B, 2009, 113, 8776 CrossRef CAS.
- N. Aumsuwan, S. Heinhorst and M. W. Urban, Biomacromolecules, 2007, 8, 713 CrossRef CAS.
- H. Kim and M. W. Urban, Langmuir, 1998, 14, 7235 CrossRef CAS.
- E. L. Florin, V. T. Moy and H. E. Gaub, Science, 1994, 264, 415 CAS.
- J. B. Huang and M. W. Urban, Appl. Spectrosc., 1992, 46(11), 1666 CrossRef CAS.
- M. W. Urban, ATR Spectroscopy of Polymers; Theory and Practice, American Chemical Society, Washington, DC, 1996 Search PubMed.
- E. Pretsch, P. Buhlmann and C. Affolter, Structure Determination of Organic Compounds: Tables of Spectral Data, Springer, New York, 2000 Search PubMed.
- N. Aumuswan, S.-H. Ye, W. R. Wagner and M. W. Urban, Langmuir, 2011, 27, 11106 CrossRef.
- A. Ebner, F. Kienberger, G. Kada, C. M. Stroh, M. Geretschlager, A. S. M. Kamruzzahan, L. Wildling, W. T. Johnson, B. Ashcroft, J. Nelson, S. M. Lindsay, H. J. Gruber and P. Hinterdorfer, ChemPhysChem, 2005, 6, 897 CrossRef CAS.
- P. Hinterdorfer and Y. F. Dufrene, Nat. Methods, 2006, 3, 347 CrossRef CAS.
- W. R. Premasiri, J. Lee and L. D. Ziegler, J. Phys. Chem. B, 2012, 116, 9376 CrossRef CAS.
Footnote |
| † Partially performed at the University of Southern Mississippi. |
| This journal is © The Royal Society of Chemistry 2013 |