Microwave-assisted rapid synthesis of poly(glycerol-sebacate) elastomers
H. M.
Aydin
*a,
K.
Salimi
b,
Z. M. O.
Rzayev
c and
E.
Pişkin
ab
aInstitute of Science, Bioengineering Division, Hacettepe University, Beytepe, 06800, Ankara, Turkey. E-mail: hmaydin@hacettepe.edu.tr; Fax: +90 312 299 21 24; Tel: +90 533 551 58 30
bChemical Engineering Department and Bioengineering Division, Centre for Bioengineering and Biyomedtek, Hacettepe University, Beytepe, 06800, Ankara, Turkey
cInstitute of Science, Nanotechnology and Nanomedicine Division, Hacettepe University, Beytepe, 06800, Ankara, Turkey
First published on 25th February 2013
Abstract
Poly(glycerol-sebacate) (PGS) was introduced a decade ago as a potential material for soft tissue repair. All of the proposed copolymerization reactions in the literature include a two-stage (prepolymerization and curing) synthesis where the reaction times can take as long as several days. This study, on the other hand, proposes a new route that eliminates these disadvantages and enables a rapid synthesis of PGS elastomers via microwave-assisted prepolymerization in minutes instead of days. No purge gas, catalyst or vacuum is needed in the first prepolymerization step. The curing stage was carried out at 150 °C for 4, 8, 16, and 24 hours. The glass transition temperature (Tg) and melting temperatures for the glycerol and sebacic acid fragments (Tm1 and Tm2) of these PGS elastomers were found as −35.61 °C, −15.82 °C, and 61.70 °C, respectively. The Young's modulus and tensile strength values were found as 0.50 ± 0.02 MPa and 0.27 ± 0.06 MPa, respectively.
1. Introduction
Biodegradable elastomers have been an important area of research in the biomedical field, from drug delivery to tissue engineering of soft tissues, and have now been replacing non-degradable elastomers thanks to their desired mechanical properties and degradation behaviour.1,2 The main advantage of these elastomers is that they do not require removal following implantation, in the case of being used as the biomaterial for the repair of a soft tissue.Poly(polyol-sebacate) elastomers, especially poly(glycerol-sebacate) (PGS), have been introduced recently and have already resulted in a large number of publications in recent years, mostly in the biomedical area. Sebacic acid and glycerol can both undergo metabolic activities in the human body with no cytotoxic effects.3 PGS elastomers can be synthesized by polycondensation of glycerol's hydroxyl groups and sebacic acid's carboxylic acid groups without the need for a catalyst.4,5 Similar polymeric materials including glycerol and sebacic acid units have been used for biomaterials applications6 with reported biocompatibility and biodegradability. PGS elastomers can also be prepared with the desired functionalities for the intended use by controlling the curing parameters. Major research areas of PGS applications in the last decade include tissue engineering of a diverse range of soft tissues such as cartilage, cardiac muscles, nerves, tympanic membranes and blood vessels.7–11
Both small and large-scale productions of such elastomers are of importance for medical and green-packaging applications; however the reaction routes proposed in the literature suggest a long and energy-consuming two-stage synthesis. A prepolymer is usually synthesized at the first stage of PGS production, followed by a curing stage of this prepolymer at elevated temperatures in vacuo. The prepolymer synthesis requires lengthy reaction times up to several days with a great deal of energy consumption for heating of the reaction mixture and applying a vacuum. Also, a large amount of purge gas, such as nitrogen, is required throughout the reaction. This route is an inefficient and unfeasible method in terms of economic considerations. Even though the curing stage cannot be altered much since the time and temperature parameters influence the final properties of the target products, this prepolymer synthesis stage can be shortened using microwave irradiation. Therefore the drawback of conventional methods, i.e. excessive energy and gas consumption, could be eliminated. In industry, microwave irradiation has been used not only in drying processes but also in chemical reactions due to its advantages over conventional heating, including the non-contact nature of the process, as well as instant and rapid heating resulting in a homogenous reaction mixture.12
Microwave polymerization has become a very popular branch of polymer science recently thanks to its advantages. The main effect of a microwave-assisted polymerization approach on the reaction speed is favorable in many reactions, especially in conventional methods requiring heating the reaction vessel for prolonged times. In addition to the outcomes such as thermal effects along with shortened reaction times, the minimization of side reactions and obtaining better quality in terms of purity makes microwave-assisted polymerization studies interesting. Of course, microwave irradiation also has several disadvantages such as a lack of information on the reaction conditions in the case of using household microwave ovens, in addition to difficulties in the control of the reaction.13
There have been number of microwave-assisted synthesis routes proposed for organic and inorganic compounds in the literature, enabling high yields because of the rapid and efficient heating of reactants.13–16 This study shows a novel synthesis route for the production of a PGS prepolymer via microwave irradiation. The method presented here enables rapid and economic production of these valuable elastomers.
2. Materials and methods
2.1. Synthesis of poly(glycerol-sebacate) (PGS) elastomers
Glycerol and sebacic acid monomers (Sigma-Aldrich) were used as received. Polymerization was performed in two stages: (i) prepolymerization, and (ii) curing. The prepolymerization stage was performed using a domestic oven. Equimolar amounts (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) of glycerol and sebacic acid monomers were weighed and loaded into Petri dishes, then transferred into a microwave oven (White Westinghouse, USA) operated at 650 W. Microwave irradiation was applied intermittently for 3 minutes with 10 second intervals, where vapor formed was removed by opening the door of the oven. No catalyst or any other reactant was used in this stage. Subsequently, these prepolymers were directly transferred to a vacuum oven (Thermo Scientific, USA) and the curing reactions were performed at 150 °C in vacuo (5 Torr) for 4, 8, 16, and 24 hours. After cooling to room temperature, solidified PGS sheets were collected and characterized. For comparison, a batch of elastomers was synthesized by using a conventional method4 under N2 atmosphere at 120 °C for 48 hours (prepolymerization) followed by curing at 140 °C in vacuo.
:1) of glycerol and sebacic acid monomers were weighed and loaded into Petri dishes, then transferred into a microwave oven (White Westinghouse, USA) operated at 650 W. Microwave irradiation was applied intermittently for 3 minutes with 10 second intervals, where vapor formed was removed by opening the door of the oven. No catalyst or any other reactant was used in this stage. Subsequently, these prepolymers were directly transferred to a vacuum oven (Thermo Scientific, USA) and the curing reactions were performed at 150 °C in vacuo (5 Torr) for 4, 8, 16, and 24 hours. After cooling to room temperature, solidified PGS sheets were collected and characterized. For comparison, a batch of elastomers was synthesized by using a conventional method4 under N2 atmosphere at 120 °C for 48 hours (prepolymerization) followed by curing at 140 °C in vacuo.
2.2. Characterization
In order to characterize the PGS elastomers and confirm polymerization, several spectroscopic and thermal analysis methods (hydrogen-nuclear magnetic resonance (1H-NMR), Fourier transform infrared spectroscopy (FTIR), and differential scanning calorimetry (DSC)) were used. The 1H-NMR spectrum was acquired using a Bruker Spectrospin Avance Ultrashield 400 spectrometer (Germany), operated at 400 MHz. The PGS elastomer samples were dissolved in CDCl3 (Sigma, 4 mg mL−1) and TMS (tetramethylsilane) was used as a reference. FTIR analysis was performed in the range 4000–400 cm−1 in a spectrophotometer (Thermo Scientific, USA), while thermal transitions were investigated using a DSC (Perkin Elmer Diamond, USA) operated at a heating rate of 10 °C min−1, under a nitrogen gas flow and the thermograms were collected from −60 °C to 300 °C. Scanning electron microscopy (SEM, Fei Nova NanoSEM 430, USA) was used to analyze the surface morphology of the prepared PGS sheets. The PGS elastomeric sample sheets for SEM were coated with Au–Pd to enhance the conductivity. End group analysis was performed by titration of the elastomer solution (0.4 g elastomer in a 0.05 M KOH titrant including an indicator) in order to calculate the number average molecular weight of the synthesized elastomers.17 The volume of KOH consumed was recorded for the elastomers and the acid number, end group concentration, and finally number average molecular weights were calculated for the elastomers cured for 4, 8, 16, and 24 hours using the formulas given in the results section. The mechanical properties of the synthesized elastomers were also investigated by tensile tests, using a Universal Tester (Lloyd, UK) with a crosshead speed of 10 mm min−1 under a 500 MPa load. Six measurements were performed and the mean values are given.3. Results and discussion
3.1. Microwave-assisted PGS synthesis
PGS elastomer synthesis was composed of two stages: prepolymerization and curing. The first stage of prepolymer formation was performed via microwave irradiation in 3 minutes. This stage was performed intermittently; the door of the domestic oven was opened for 10 seconds in every minute in order to remove the vapor formed due to the polycondensation reaction. Immediately after the prepolymerization stage, elastomers were subjected to curing for different periods of time (4, 8, 16, and 24 hours). Curing times of 4 and 8 hours resulted in a wax-like structure with a poor film-forming capacity, while the elastomers cured for 16 and 24 hours gave elastic sheets with good mechanical integrity. As a result, the data presented in this section are for the elastomers cured for 16 hours. The curing stage was performed at a relatively high temperature (i.e. 150 °C) and therefore this observation is dependent on the curing times. The molecular structure of the synthesized elastomer includes hydrogen bonds and cross-linking between the carboxylic acid groups and the hydroxyl groups. Moreover there were some intra- and inter-molecular interactions present. The molecular structure also allows for branching, mainly from the central –OR moiety. Yet, cross-links are formed between backbones and available grafting chains, and the number of these cross-links is found to be correlated to the curing temperature and time. The higher the curing temperature, hydroxyl–hydroxyl condensation reactions are more likely to occur.3.2. Chemical characterization
Polymerization was also confirmed by 1H-NMR analysis (Fig. 1). Calculations on the integration of peak areas were performed to reveal the monomer ratios of the final product. The method used for these calculations is a modified version of the known 1H-NMR method.18 It is worth to note that the monomer feed ratio was selected as 1:1 (n/n). Comparing the integral areas of the respective protons from the –(CH2)2– (C2 and C7) group of the m1 unit and the CH (C10) group of the m2 unit regions in the spectra of the copolymer resulted in the following eqn (1):| m1/m2 = n2Am1/n1Am2 = 3.559 | (1) |
| m |
| 1 |
| (sebacic acid): |
| m |
| 2 |
| (glycerol) = 78:22. |
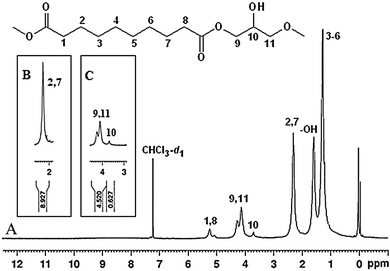 | ||
| Fig. 1 1H-NMR spectrum (A) of poly(glycerol-sebacate) elastomers with related peak regions (B and C) used to calculate the monomer ratios. | ||
Since the main advantage of microwave polymerization is that the energy can be distributed rapidly and homogeneously throughout the reaction mixture,18 and this eventually leads to a faster polymerization; the shorter reaction times in the present study are not due to the higher temperature levels. Because, elevated temperatures might easily decompose the monomers or at least boil off the monomer with a lower boiling point. The boiling points for glycerol and sebacic acid are 290 °C and 374 °C (at 760 mmHg), respectively. Since the boiling point of the sebacic acid monomer is higher than that of glycerol, this may lead to boiling off of some of the glycerol monomers in the microwave and vacuum stages (curing). Moreover, in a closed reaction system, a 1:1 (n/n) monomer ratio leads to the formation of block sebacic acid fragments when the glycerol monomers are purged or boiled off; however, in here, this is highly unlikely to occur since the boiled off fractions might leave the system.
Fig. 2 below shows the differences between the spectra gathered from conventionally produced (a) and microwave-irradiated (b) samples. It is seen from the figure that the peaks that appeared in the areas corresponding to the formation of C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds at 1159 and 1735 cm−1 were not similar between the two prepolymers, even though the other peak groups were found to be the same.
O bonds at 1159 and 1735 cm−1 were not similar between the two prepolymers, even though the other peak groups were found to be the same.
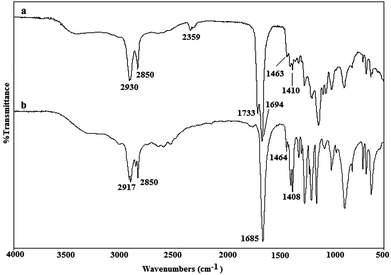 | ||
| Fig. 2 Fourier transform infrared (FTIR) spectra of poly(glycerol-sebacate) prepolymers synthesized via (a) a conventional reaction at 120 °C for 48 hours under N2, and (b) 3-minutes of microwave irradiation (650 W). | ||
Fig. 3(A) shows the Fourier transform infrared spectrum of the elastomers. The peaks at 2927–2852 cm−1 were attributed to alkene (–CH2) groups, while intense peaks at 1159 and 1735 cm−1 were due to the formation of C–O and CO, respectively. Absorption of methyl groups (–CH3 bending) also appeared at 1354–1456 cm−1. The O–H stretch peak was shown at 935 cm−1 while the peak at 1416 cm−1 was attributed to the O–H bending mode. The spectra in Fig. 3(B) correspond to elastomer samples cured for 4, 8, 16, and 24 hours following microwave irradiation of 3 minutes while spectrum ‘e’ corresponds to that synthesized via the conventional method (24 hours prepolymerization followed by a 5-day curing at 140 °C in a vacuum). It is shown that the peaks at 1704–1707 cm−1 reduced their intensity when the curing time increases. The peaks at 1734 cm−1 were sharpened upon increased curing time. This is an indication of the formation of cross-linking. On the other hand, the spectrum for the 16 h-cured sample was the closest fit to the conventionally produced sample in terms of the degree of cross-linking.
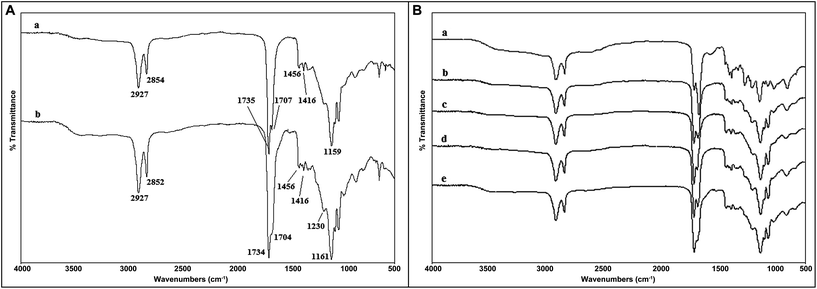 | ||
| Fig. 3 (A) Fourier transform infrared (FTIR) spectra of the poly(glycerol-sebacate) elastomers synthesized via (a) conventional reaction (48 hours) followed by 5-days of curing, and (b) 3-minutes of microwave irradiation (650 W) followed by 16 h of curing under N2; and (B) Fourier transform infrared (FTIR) spectra of the poly(glycerol-sebacate) elastomers synthesized via microwave irradiation followed by curing times of 4, 8, 16, and 24 hours (a, b, c, and d, respectively), and (e) conventional reaction (48 hours) followed by 5-days of curing. | ||
Typical thermal transitions of the synthesized elastomers were calculated by DSC. The thermogram given in Fig. 4(A) suggests that these PGS elastomers are semicrystalline below the melting temperature (Tm) and amorphous at 37 °C. In the literature, PGS elastomers were reported to have a glass transition temperature (Tg) of −37.02 °C19 and Tm values of 5.23 °C and 37.62 °C for sebacic acid and glycerol moieties, respectively. From Fig. 4(A), the PGS elastomers synthesized in this study (microwave assisted, 16 h curing time) have a Tg of −35.61 °C (ΔCp = 0.047 J g−1 °C−1). Two melting temperatures (Tm) for the microwave-assisted synthesized PGS elastomers were found as Tm1 = −15.82 °C (ΔCp = 0.340 J g−1 °C−1), and Tm2 = 61.70 °C (ΔCp = 0.065 J g−1 °C−1), corresponding to glycerol and sebacic acid fragments, respectively. Please note that the differences in Tm values are stemmed from the final monomer ratio calculated here as 78:22 for sebacic acid:glycerol monomers. Fig. 4(B) shows the DSC thermograms of the elastomers synthesized via 3 minutes microwave irradiation followed by 8 h (a), 16 h (b), and 24 h (c) curing stages, as well as that prepared via a conventional method (d) (48 hours of the first stage followed by a 5-day curing stage). A shift in Tm1 is observed, starting from the samples cured for 8 hours to 24 hours following microwave irradiation when compared to that prepared by the conventional method. The sample cured for 4 hours did not give a thermogram and therefore it is not included in the figure.
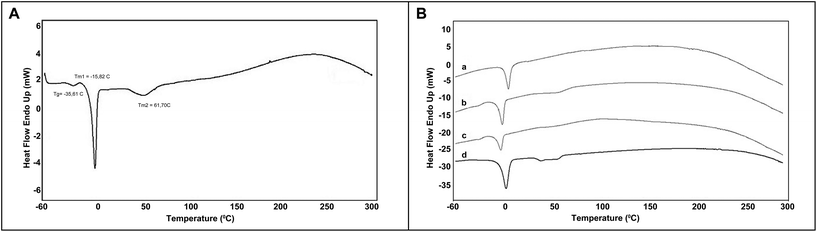 | ||
| Fig. 4 (A) A representative DSC thermogram of poly(glycerol-sebacate) elastomers synthesized via a microwave-assisted two stage method (curing time of 16 hours); and (B) DSC thermograms of the elastomers synthesized via microwave irradiation followed by curing times of 8, 16, and 24 hours (a, b, and c, respectively), and (d) conventional reaction (48 hours) followed by 5-days of curing. | ||
3.3. Mechanical properties
The Young's modulus of PGS elastomers reported in the literature was in the range 0.05–1.5 MPa.20 Tensile tests (graph shown in Fig. 5) showed that the PGS elastomers synthesized in this study have a Young's modulus of 0.50 ± 0.02 MPa, tensile strength of 0.27 ± 0.06 MPa, and a percentage of elongation of approximately 180%. These results are consistent with the values from the literature.20,21 However, other publications, of course, may report different values since this property is directly correlated to several parameters such as the molecular weight, cross-linking density and the final monomer composition of the elastomer.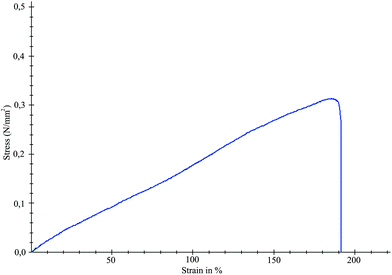 | ||
| Fig. 5 Tensile test graph of poly(glycerol-sebacate) elastomers (microwave stage of 3 minutes and curing time of 16 hours). | ||
SEM images were collected for the investigation of the surface morphology of the prepared sheets (a representative image is given in Fig. 6). A rough topography was observed. It should be noted that the SEM image was obtained from the outer surface of the cast film, which was taken directly out of the vacuum oven after the curing reaction. Also, the whole reaction (both stages) was performed in a Petri dish; therefore this is yet another advantage of the present method since it eliminates the use of glassware such as 3-neck glass reactors etc. for the reaction. The SEM image is given purely to provide additional information regarding the morphology for the readers who would like to observe the surface topography since the prepared elastomers are ready to go after polymerization thanks to the intrinsic mechanism of the present method which excludes the use of any catalyst or solvent.
 | ||
| Fig. 6 A surface SEM image of poly(glycerol-sebacate) (microwave stage of 3 minutes and curing time of 16 hours). | ||
In order to calculate the acid number (AN, mgKOH g−1) value, titrations were performed to calculate the amount of KOH consumed (V(KOH)) according to the following equation:
| AN = [Mw(KOH)·V(KOH)·N(KOH)]/m | (2) |
The acid number values were then put into eqn (3) to calculate the carboxylic acid end group concentration (EGC, %):
| EGC = [(AN) × (Mw(COOH))]/Mw(KOH) | (3) |
Eventually, the number-average molecular weights of the synthesized elastomers were calculated by eqn (4):
| Mn = [106/(EGC)] | (4) |
Table 1 summarizes the amount of KOH consumed, acid numbers, end group (–COOH) concentration values, and number average molecular weights for the elastomers cured for 4, 8, 16, and 24 hours following a 3 minutes microwave-irradiation stage. It was found that the film properties of the elastomers cured for 4 and 8 hours were insufficient in terms of integrity.
| Cure time (hours) | KOH consumed (mL) | Acid number (mgKOH g−1) | End group concentration (%) | Number average molecular weight (kDa) |
|---|---|---|---|---|
| 4 | 8.5 | 59.60 | 47.80 | 20.92 |
| 8 | 5.0 | 35.06 | 28.12 | 35.46 |
| 16 | 2.4 | 16.83 | 13.49 | 74.12 |
| 24 | 2.2 | 15.67 | 12.56 | 79.61 |
4. Conclusion
In this study, we report a rapid production method for the synthesis of an important biodegradable elastomer, PGS via microwave irradiation. It was shown that microwave irradiation could also provide the heat energy required for the prepolymerization reaction without the need for a vacuum atmosphere. Neither a catalyst nor purge gas was used in the reaction.A polycondensation reaction between the hydroxyl end groups of the glycerol and carboxylic acid end groups of the sebacic acid monomers via this microwave assisted synthesis route enabled the formation of prepolymers in less than 5 minutes, instead of reported reaction times up to several days for the same stage. The curing stage in the synthesis could also be decreased down to 16 hours following the microwave stage instead of reported times between 2 to 5 days. Therefore, the method given in this study is highly advantageous in terms of economical concerns especially for the bulk production of such elastomers.
However, this microwave-assisted reaction yields a polymer with a different molar ratio from the starting molar composition (100:100 to 78:22). This may be due partly to the reaction temperature during the first prepolymerization step via boiling off of the glycerol monomers as well as higher curing temperatures in the second stage. Given that the boiling point of glycerol is 290 °C, it is possible that the decreased time needed for polymerization is indeed caused by much higher temperatures. Regardless of the exact mechanism underlying this observation, it is important to emphasize that the final monomer ratio of 78:22 (at the end of curing stage) may be altered by playing with the reaction times and temperatures. Of course, since this elastomer differs in the final ratio than previously reported ones with 1:1 ratios, it is understood that the given FTIR and DSC data are slightly different. Still, this material with 78:22 ratios can find diverse applications in biomaterials and environmentally friendly applications such as green plastics.
The method for the production of PGS elastomers given in this study can also be scaled up since these elastomers have an enormous importance not only in biomedical areas but also in engineering plastics fields such as packaging and green plastic applications. Scaling up the production approach of this study may result in a rapid and economic manufacturing of these elastomers. In 2009, the first commercial plant for the mass production of poly(lactic acid) via a microwave method has been developed in Japan.
Current production methods are limited in terms of reaction conditions, i.e. purging an expensive gas (such as nitrogen) for days and conventionally heating and stirring the reaction mixture. Even though there have been several reports regarding the production of PGS elastomers in shorter times up to 120 minutes (e.g.ref. 22), most of these faster approaches suggest the use of a catalyst. A catalyst is not recruited in this study since the final application requires pure elastomers. Recent catalyst-free methods report total polymerization times of up to one week. Therefore, it is for the first time that PGS elastomers are synthesized via a microwave-assisted approach and the reaction times reduce from days to minutes in the prepolymerization step and the total reaction times are approximately reduced by more than 80%. In addition to the future industrial applications of these elastomers, now researchers in the biomaterials area can readily synthesize and modify these important elastomers for diverse applications.
Acknowledgements
Chemicals were kindly donated by BMT Calsis Co., Turkey. Erhan Pişkin is supported by the Turkish Academy of Sciences as a full member.References
- J. P. Bruggeman, B. J. de Bruin, C. J. Bettinger and R. Langer, Biodegradable poly(polyol sebacate) polymers, Biomaterials, 2008, 29(36), 4726–4735 CrossRef CAS.
- Q. Y. Liu, L. Jiang, R. Shi and L. Q. Zhang, Synthesis, preparation, in vitro degradation, and application of novel degradable bioelastomers – a review, Prog. Polym. Sci., 2012, 37(5), 715–765 CrossRef CAS.
- D. G. Barrett and M. N. Yousaf, Design and applications of biodegradable polyester tissue scaffolds based on endogenous monomers found in human metabolism, Molecules, 2009, 14(10), 4022–4050 CrossRef CAS.
- Y. Wang, G. A. Ameer, B. J. Sheppard and R. Langer, A tough biodegradable elastomer, Nat. Biotechnol., 2002, 20(6), 602–606 CrossRef CAS.
- Q. Y. Liu, M. Tian, T. Ding, R. Shi, Y. X. Feng and L. Q. Zhang, et al., Preparation and characterization of a thermoplastic poly(glycerol sebacate) elastomer by two-step method, J. Appl. Polym. Sci., 2007, 103(3), 1412–1419 CrossRef CAS.
- J. Fu, J. Fiegel, E. Krauland and J. Hanes, New polymeric carriers for controlled drug delivery following inhalation or injection, Biomaterials, 2002, 23(22), 4425–4433 CrossRef CAS.
- C. G. Jeong and S. J. Hollister, A comparison of the influence of material on in vitro cartilage tissue engineering with PCL, PGS, and POC 3D scaffold architecture seeded with chondrocytes, Biomaterials, 2010, 31(15), 4304–4312 CrossRef CAS.
- Q. Z. Chen, H. Ishii, G. A. Thouas, A. R. Lyon, J. S. Wright and J. J. Blaker, et al., An elastomeric patch derived from poly(glycerol sebacate) for delivery of embryonic stem cells to the heart, Biomaterials, 2010, 31(14), 3885–3893 CrossRef CAS.
- C. A. Sundback, J. Y. Shyu, Y. Wang, W. C. Faquin, R. S. Langer and J. P. Vacanti, et al., Biocompatibility analysis of poly(glycerol sebacate) as a nerve guide material, Biomaterials, 2005, 26(27), 5454–5464 CrossRef CAS.
- A. M. Wieland, C. A. Sundback, A. Hart, K. Kulig, P. T. Masiakos and C. J. Hartnick, Poly(glycerol sebacate)-engineered plugs to repair chronic tympanic membrane perforations in a chinchilla model, Otolaryngol.–Head. Neck Surg., 2010, 143(1), 127–133 CrossRef.
- D. Motlagh, J. Yang, K. Y. Lui, A. R. Webb and G. A. Ameer, Hemocompatibility evaluation of poly(glycerol-sebacate) in vitro for vascular tissue engineering, Biomaterials, 2006, 27(24), 4315–4324 CrossRef CAS.
- D. Bogdal, P. Penczek, J. Pielichowski and A. Prociak, Microwave assisted synthesis, crosslinking, and processing of polymeric materials, Adv. Polym. Sci., 2003, 163, 193–263 CrossRef CAS.
- F. Wiesbrock, R. Hoogenboom and U. S. Schubert, Microwave-assisted polymer synthesis: state-of-the-art and future perspectives, Macromol. Rapid Commun., 2004, 25(20), 1739–1764 CrossRef CAS.
- S. Mishra, G. U. Rani and G. Sen, Microwave initiated synthesis and application of polyacrylic acid grafted carboxymethyl cellulose, Carbohydr. Polym., 2012, 87(3), 2255–2262 CrossRef CAS.
- P. Rani, G. Sen, S. Mishra and U. Jha, Microwave assisted synthesis of polyacrylamide grafted gum ghatti and its application as flocculant, Carbohydr. Polym., 2012, 89(1), 275–281 CrossRef CAS.
- X. G. Huang, X. L. Jiang and R. X. Zhuo, Microwave-assisted solid-phase synthesis of pH-responsive polyaspartamide derivatives, Carbohydr. Polym., 2012, 89(3), 788–794 CrossRef CAS.
- J. F. Rabek, Experimental Methods in Polymer Chemistry, John Wiley and Sons, New York, 1980 Search PubMed.
- N. R. Cameron, J. M. G. Cowie, R. Ferguson and I. McEwan, Enthalpy relaxation of styrene-maleic anhydride (SMA) copolymers Part 1. Single component systems, Polymer, 2000, 41(19), 7255–7262 CrossRef CAS.
- W. Cai and L. L. Liu, Shape-memory effect of poly (glycerol-sebacate) elastomer, Mater. Lett., 2008, 62(14), 2171–2173 CrossRef.
- Q. Z. Chen, A. Bismarck, U. Hansen, S. Junaid, M. Q. Tran and S. E. Harding, et al., Characterisation of a soft elastomer poly(glycerol sebacate) designed to match the mechanical properties of myocardial tissue, Biomaterials, 2008, 29(1), 47–57 CrossRef CAS.
- I. H. Jaafar, M. M. Ammar, S. S. Jedlicka, R. A. Pearson and J. P. Coulter, Spectroscopic evaluation, thermal, and thermomechanical characterization of poly(glycerol-sebacate) with variations in curing temperatures and durations, J. Mater. Sci., 2010, 45(9), 2525–2529 CrossRef CAS.
- M. Nagata, T. Kiyotsukuri, H. Ibuki, N. Tsutsumi and W. Sakai, Synthesis and enzymatic degradation of regular network aliphatic polyesters, React. Funct. Polym., 1996, 30(1–3), 165–171 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |