PR_b functionalized stealth liposomes for targeted delivery to metastatic colon cancer†
Maroof
Adil
a,
Lalitha
Belur
b,
Timothy R.
Pearce
c,
Rachel M.
Levine
a,
Alison W.
Tisdale
a,
Brent S.
Sorenson
d,
R. Scott
McIvor
b and
Efrosini
Kokkoli
*a
aDepartment of Chemical Engineering and Materials Science, University of Minnesota, Minneapolis, MN 55455, USA. E-mail: kokkoli@umn.edu; Fax: +1 (612) 626-7246; Tel: +1 (612) 626-1185
bDepartment of Genetics, Cell Biology and Development, University of Minnesota, Minneapolis, MN 55455, USA
cDepartment of Biomedical Engineering, University of Minnesota, Minneapolis, MN 55455, USA
dDepartment of Diagnostic and Biological Sciences, University of Minnesota, Minneapolis, MN 55455, USA
First published on 13th December 2012
Abstract
A gene delivery system was designed to carry a payload to integrin overexpressing cells. Branched-polyethyleneimine (bPEI) condensed plasmid DNA was encapsulated into targeted stealth liposomes, thereby combining the condensing and transfection properties of bPEI with the stealth and targeting properties of the liposomal carrier system. PR_b was used as a targeting ligand – a peptide we designed to bind specifically to the cancer cell surface marker α5β1 integrin – and such a robust receptor–ligand interaction achieved higher specificity than what has been previously reported for targeted delivery systems. In the process of formulating the PR_b functionalized gene delivery vehicle, we developed a protocol to fully encapsulate condensed DNA in liposomes and accurately quantify the total DNA in the system. We demonstrate that compared to non-targeted stealth liposomes and non-encapsulated condensed DNA, the PR_b functionalized stealth liposomes mediated improved in vitro transfection specifically to colon cancer cells overexpressing the α5β1 integrin. Furthermore, when administered in vivo to metastatic tumor bearing mice, PR_b functionalized stealth liposomes outperformed non-targeted liposomes and delivered genes specifically to the tumor site.
1. Introduction
Advances in genetics have made gene therapy an increasingly plausible method of disease treatment, and the search for better and more efficient gene delivery techniques continues. Free plasmid DNA faces many challenges in gene therapy in vivo: it is rapidly degraded by enzymes in the blood,1 it is limited by low levels of cellular uptake and subsequently poor intracellular release, leading to low transfection efficiencies.2 Gene delivery vehicles can overcome these issues by protecting the plasmid DNA through compaction and improving the transfection efficiency through the use of agents that increase cellular uptake and intracellular release.Both viral and non-viral methods have been used for delivery of therapeutic genes,3–8 the latter being advantageous due to reduced immunogenicity, lower toxicity and ease of manufacture.9–11 Non-viral vectors for DNA delivery include preparations containing cationic polymers and lipids.12–14 Out of these, polyethyleneimine (PEI) has been widely investigated and has shown much promise as a transfection agent.15–17 PEI condenses DNA into small particles and allows efficient DNA release in target cells by the “proton sponge effect”.18–20 However transfection by PEI alone has been shown to be toxic to cells.2,21 To overcome this problem, PEI has been modified in various ways to reduce its toxicity and improve transfection efficiency and biocompatibility through conjugation to stabilizing polymers or lipids,22–26 modifications of PEI linkages,27,28 and inclusion of a targeting moiety to improve specificity of delivery.29–31 PEI-DNA complexes have also been encapsulated in liposomes, or associated with liposomes to form lipopolyplexes,32–35 and such association has been shown to increase the transfection efficiency of PEI-condensed DNA systems.36
Encapsulating payloads in liposomes holds several advantages over conventional administration of free therapeutics. Liposomes with diameters of 100–200 nm can specifically accumulate in tumor tissue by a passive targeting mechanism involving the enhanced permeability and retention (EPR) effect.37,38 In addition, the steric effect of a polyethylene glycol (PEG) brush layer on stealth liposomes helps to avoid detection by the reticuloendothelial system (RES)39,40 and prolongs circulation lifetime. Briefly, liposomes in this size range are able to permeate through the larger membrane fenestrations in tumor blood vessels into the tumor tissue. Liposomes much larger may non specifically accumulate in the spleen by a filtration mechanism, or be trapped in capillaries in the lung, while smaller particles can non specifically permeate into other tissues or experience ineffective steric hindrance leading to a reduced circulation lifetime.38,41–43 In order to further increase the specificity of the delivery system, various targeting moieties can be incorporated into the basic carrier system. Targeted liposomes have been used in the delivery of payloads to various disease sites including tumors.44–47 However the benefits of targeting remain controversial. Although targeted liposomes reduce non specific delivery compared to the free drug, there remains considerable room for improvement. When administered in vivo, targeted delivery vehicles often accumulate to significant levels in healthy tissues,46,48–50 potentially causing undesirable toxic side effects.
Identification of an optimal receptor–ligand pair that allows the delivery vehicle to interact with diseased cells expressing high levels of the receptor, and not to healthy cells expressing the receptor at a lower concentration, can lead to improved specificities of therapeutic delivery. α5β1 integrin is a promising target that has been shown to be overexpressed on both cancer cells as well as on cancer vasculature, and minimally expressed on normal healthy tissues,51–55 and associated with increased tumorigenicity, malignancy and tumor cell invasiveness.51,56,57 In addition, α5β1 is also expressed on certain primary cell lines, but its expression is downregulated during development.58 RGD is a peptide that binds integrins including α5β1 integrin and is probably one of the most widely studied ligands in targeted drug delivery.45,46,48–50,59–62 However, RGD targeted delivery systems do not reduce accumulation in healthy organs when administered in vivo.46,48–50 Hence there is the need to develop better targeting systems capable of achieving increased in vivo specificity. Our group has previously designed a targeting peptide PR_b based on fibronectin, the native binding ligand for α5β1 integrin by combining the RGD binding site and the PHSRN synergy site. This combination of sites is specific for binding to α5β1.63,64 PR_b has outperformed both fibronectin and RGD in terms of binding cells expressing the α5β1 integrin.63 PR_b functionalized vehicles previously formulated in our group have shown increased in vitro delivery of model payloads when compared to non-targeted vehicles.65–69 In this study, we investigate the potential of the PR_b targeted liposomal system as a new transfection agent in vitro, and for the first time test its functionality in vivo.
To date, there has been a minimal amount of work reported on encapsulating condensed plasmid DNA into a targeted liposome and verifying specificity of the delivery vehicle. This is most likely due to two factors. First is the difficulty of encapsulating a large, condensed plasmid DNA particle into the limited internal volume of a 100–200 nm liposome. Second, the proximity in size of unencapsulated particles and condensed DNA containing liposomes presents a challenge in their separation and the quantification of total DNA in the final preparation. This paper presents a DNA encapsulated liposome formulation procedure that addresses each of these challenges.
In this study we encapsulated condensed DNA into PR_b functionalized stealth liposomes. We subsequently purified the system and quantified total DNA to formulate a vehicle that delivers the desired DNA load specifically to cells bearing the α5β1 integrin. We have investigated the ability of cationic polymers poly-L-lysine (pLL) and branched polyethyleneimine (bPEI) to condense DNA into small particles that can be encapsulated into a 200 nm liposome. We also tested the in vitro DNA transfection efficiency of these condensing agents in both liposomal and non liposomal forms. We used the most efficient condensing system, bPEI in our case, to optimize the concentration of the PR_b targeting moiety on targeted stealth liposomes. We demonstrate the superior performance by the PR_b functionalized gene delivery system when compared to non-targeted liposomes, or unencapsulated condensed DNA, in terms of transfection efficiency and targeting specificity in vitro and in a metastatic tumor model in vivo.
2. Materials and methods
2.1 DNA condensation
20 μg of plasmid pT2/Cal,70 which encodes the firefly luciferase transgene, under transcriptional control of a chimeric CAGS promoter (CMV enhancer/chicken beta-actin promoter/chicken beta-globin intron sequence) was used in this study. An endotoxin-free stock solution of this plasmid was diluted to 125 μl in distilled water. The amine to phosphate (N/P) ratio of the DNA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :polymer mixture was calculated as shown previously.71 Calculated volumes of the condensing agent 22.5 kDa pLL (Sigma Aldrich, Saint Louis, MO) or 25 kDa bPEI (Sigma Aldrich, Saint Louis, MO) to achieve a particular N/P ratio were also diluted to 125 μl in distilled water. The condensing agent was then added rapidly to the DNA solution followed by rapid pipetting to allow quick mixing. The system was then vortexed at the lowest speed for 5 minutes and then incubated at room temperature for 30 minutes. DNA particle size was measured by dynamic light scattering (DLS) (Brookhaven Instruments Corporation) and zeta potential determined using the PALS zeta potential analyzer (Brookhaven Instruments Corporation).
:polymer mixture was calculated as shown previously.71 Calculated volumes of the condensing agent 22.5 kDa pLL (Sigma Aldrich, Saint Louis, MO) or 25 kDa bPEI (Sigma Aldrich, Saint Louis, MO) to achieve a particular N/P ratio were also diluted to 125 μl in distilled water. The condensing agent was then added rapidly to the DNA solution followed by rapid pipetting to allow quick mixing. The system was then vortexed at the lowest speed for 5 minutes and then incubated at room temperature for 30 minutes. DNA particle size was measured by dynamic light scattering (DLS) (Brookhaven Instruments Corporation) and zeta potential determined using the PALS zeta potential analyzer (Brookhaven Instruments Corporation).
2.2 DNA liposome formulation and characterization
1,2-Dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-dipa-lmitoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(poly-ethylene glycol)-2000] (ammonium salt) (DPPE-PEG2000) and cholesterol were purchased from Avantipolar Lipids, Inc. (Alabaster, AL). Non-targeted stealth liposomes were made with 55 mol% DPPC, 35% cholesterol and 5% DPPE-PEG2000. Targeted stealth liposomes were made with (55 − x)% DPPC, 35% cholesterol, 5% DPPE-PEG2000 and x% PR_b peptide amphiphile, where x was varied between 1.5 and 9. The PR_b peptide was purchased from Biomedical Genomics Center (BMGC), University of Minnesota and the peptide amphiphile was made as described previously.63,72 All liposomes were made using the thin film hydration method,73 where the mixture of lipids was deposited on the bottom of a 50 ml round bottom flask and dried under a stream of argon followed by overnight incubation in a vacuum oven. The films were then hydrated with a solution of condensed DNA in DI water and incubated at 45–50 °C in a water bath for 2 hours. The resulting liposomes were extruded through 200 or 400 nm polycarbonate membranes (Avestin, Ottawa, Canada) 11 times with a handheld Liposofast extruder (Avestin, Ottawa, Canada). Liposomes were then purified by a 24 hour dialysis through a 1000 kDa cellulose ester dialysis membrane (Spectrum Labs, Rancho Dominiguez, CA). Lipid concentration was measured using a phosphorus assay as described elsewhere74 (reagents from Sigma Aldrich). PR_b concentration on the liposomes was determined by the BCA assay (Thermo Scientific, Waltham, MA) following the manufacturer's protocol. The PR_b concentration is expressed as a mole percentage of total lipid. pT2/Cal plasmid DNA was condensed using initially pLL at a N/P ratio of 4 and with bPEI at a N/P ratio of 8 (choices justified in ESI, Fig. S1†), and encapsulated in liposomes functionalized with approximately 5 mol% PR_b (actual concentrations are shown in the figure captions) or in non-targeted stealth liposomes. The ZetaPALS Zeta Potential Analyzer (Brookhaven Instruments, Holtsville, NY) was used to determine both the zeta potential, and diameter (by dynamic light scattering; DLS) of the stealth liposomes that encapsulated bPEI condensed DNA at N/P ratio of 8. To further characterize the size and shape of stealth liposomes encapsulating condensed DNA, cryogenic transmission electron microscope (cryo-TEM) images were taken. Briefly, 4 μl of liposome samples dissolved in water were deposited onto a lacey formvar/carbon copper grid (Ted Pella) treated for 15 s with glow discharge and vitrified in liquid ethane by Vitrobot (Vitrobot parameters: 5 s blot time, −1 offset, 3 s wait time, 3 s relax time, 95% humidity). Following vitrification, the grid was transferred to a Tecnai G2 Spirit TWIN 20–120 kV/LaB6 Transmission Electron Microscope. Images were captured using an Eagle 2k CCD camera with an accelerating voltage of 120 kV.2.3 DNA quantification
Plasmid DNA was labeled with cy5 fluorescent dye using the Label IT cy5 DNA labeling kit (Mirus, Madison, WI) according to the manufacturer's protocol. 5% of the plasmid DNA used in the DNA condensation process was cy5 labeled. At the post hydration stage of liposome preparation, a DNA concentration standard curve was generated, which was used to quantify the total DNA in the system after the final purification step.2.4 In vitro transfection
DLD-1 human colon carcinoma cells (ATCC, Manassas, VA) were cultured in T75 flasks using Dulbecco-modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum and penicillin–streptomycin to 70% confluence and then subcultured into 96 well plates at 5000 cells per well 24 hours prior to transfection. All cell culture reagents were purchased from the Invitrogen Supply Center, University of Minnesota. On the day of transfection media was replenished and DNA liposomes (liposomes containing 100 ng of condensed DNA) were added to wells in replicates of six and incubated at 37 °C for 40 hours. Luciferase expression was then measured with the Luciferase Reporeter assay kit (Promega, Madison, WI) following the manufacturer's protocol. Luminescence measured from untreated cells was used as the background, and that measured from 100 ng of unencapsulated DNA condensed with either pLL or bPEI under the same conditions was used as control.2.5 Effect of PR_b concentration on transfection
Different PR_b concentrations were used to prepare PR_b functionalized PEGylated liposomal films. Each of these films was hydrated using DNA condensed with bPEI at an N/P ratio of 8. DNA liposomes were then prepared and characterized as described above. DLD-1 cells subcultured in a 96 well plate were transfected using 100 ng of liposomal DNA, and luciferase activity was assayed as described above in the in vitro transfection section.2.6 Specificity of targeting
DLD-1 cells were seeded in a white 96 well plate at 5000 cells per well and cultured overnight. Cells were provided with fresh media the next day and the plate was placed at 4 °C for 30 minutes to equilibrate before the addition of 4 μg of the GRGDSP peptide (BMGC, University of Minnesota) to each well. Following 30 minutes incubation at 4 °C, bPEI-DNA encapsulated in PR_b functionalized stealth liposomes were added to each well. The plate was incubated at 37 °C and 5% CO2 for a total of 32 hours with the addition of 4 μg per well GRGDSP peptide every 8 hours to maintain a high concentration of free peptide for the purposes of blocking integrin binding. As positive controls, the same liposomes were used without GRGDSP blocking. All wells received 100 ng DNA, as quantified by the cy5 quantification assay described above. At the end of the incubation period, luciferase expression was assayed as described above. To demonstrate the binding specificity of the PR_b peptide, a scrambled PR_b sequence (ScrPR_b, SGRSGSGSHGGDSGSS-KSSPNPR), was obtained from the BMGC at the University of Minnesota, and used as an alternate targeting ligand on liposomes. DLD-1 cells plated at 5000 cells per well in a white 96 well plate were transfected with PR_b or ScrPR_b functionalized stealth liposomes. In parallel, DLD-1 cells were first blocked with 16 μg per well free PRb peptide followed by transfection with either the PR_b or ScrPR_b targeted liposomes for 32 hours at 37 °C in the presence of free PR_b added every 8 hours. All wells received 100 ng of DNA encapsulated in liposomes as quantified by the cy5 quantification assay. At the end of this incubation period, luciferase expression was measured as described above.2.7 Establishment of hepatic tumors
Female Balb/c mice were obtained from NIH (Frederick, MD) and maintained under specific pathogen-free conditions. All animals were treated according to the NIH Guidelines for Animal Care with approval of the IACUC of the University of Minnesota. Exponentially growing murine CT26 colon carcinoma cells (ATCC, Manassas, VA) were trypsinized, resuspended in Hanks balanced salt solution (HBSS), and 1 × 105 viable cells in a volume of 150 μl were injected intrasplenically into 4–6 week mice. Animals were administered an anesthetization cocktail consisting of ketamine HCl (8 mg ml−1; Phoenix Scientific, St. Joseph, MO), acepromazine maleate (0.1 mg ml−1; Phoenix Scientific), and butorphanol tartrate (0.01 mg ml−1; Fort Dodge Animal Health, Overland Park, KS). A peritoneal incision was made, and cells were directly injected into the spleens of recipient animals. Pressure was applied to the injection site for 3 minutes, and the animals were splenectomized. Cells injected into the spleen in this manner flow from the splenic vein into the portal vein and seed into the liver, thus confining tumor formation to the liver. The incision site was closed with staples, and the animals were allowed to recover.2.8 In vivo liposomal gene delivery and luciferase imaging
Animals were injected with liposomal formulations at 2 weeks post-tumor injection, with tumor growth determined by measuring the abdominal girth of the animal. The three groups investigated were tumor bearing mice that received non-targeted stealth liposomes, tumor-bearing mice that received PR_b functionalized stealth liposomes, and healthy, non-tumor bearing mice that received PR_b functionalized stealth liposomes. Four animals per group were injected intravenously via the lateral tail vein, with liposomal formulations containing 700 ng of plasmid DNA in a total volume of 0.5 ml. 24 hours after liposomal injections, expression of luciferase was visualized by in vivo bioluminescence imaging using the Xenogen IVIS Imaging System (Xenogen, Alameda, CA). Mice were anaesthetized using the anesthetization cocktail described above, followed by an intraperitoneal injection of 100 μl luciferin (Promega) and the whole mouse was imaged for bioluminescence 5 minutes later. Mice were sacrificed, livers extracted, and imaged ex vivo in 100 μl PBS containing 100 μl luciferin. Luciferase expression is reported as photons emitted per second per cm2.3. Results and discussion
3.1 Characterization of targeted and non-targeted stealth liposomes encapsulating condensed DNA
Unlike many previous gene delivery systems where polyplexes are complexed with anionic liposomes, we have encapsulated polyplexes into liposomes composed of neutral lipids. In this process, there is no active interaction between the polyplexes and the liposomes. Polyplexes in the aqueous phase are passively encapsulated into liposomes by a thin film lipid hydration process. 20 μmol lipid films were hydrated with 20 μg of cationic polymer-condensed DNA. After the purification step, where unencapsulated condensed DNA is removed, an average concentration of about 5 μg ml−1 DNA, corresponding to a 40% yield, is obtained in the final DNA–liposome formulation.Three characteristic stealth liposomes each, for PR_b functionalized and non-targeted stealth formulations, containing bPEI condensed DNA at N/P 8 were analyzed. Measured sizes were 204 ± 6 nm with zeta potential of −15.3 ± 0.6 mV for the non-targeted stealth liposomes. The size of the PR_b functionalized stealth liposomes was 279 ± 51 nm and the zeta potential was −3.1 ± 1.8 mV. Although the bPEI–DNA complexes are positively charged, once encapsulated in stealth liposomes the system as a whole is close to neutral or slightly negative. The lipid bilayer and the PEG layer effectively shield the charge of the encapsulated DNA particles. In addition, cryo-TEM images of PR_b functionalized stealth liposomes encapsulating bPEI condensed DNA were taken and shown in Fig. 1. These images confirm the presence of approximately 100 nm sized condensed DNA particles within spherical stealth liposomes sized around 250 nm, correlating with the sizes measured using DLS.
 | ||
| Fig. 1 Cryo-TEM image of 5 mol% PR_b functionalized stealth liposomes (5 mol% PEG2000) encapsulating bPEI condensed DNA. | ||
3.2 Transfection efficiency of liposomes encapsulating pLL and bPEI condensed DNA
For both pLL and bPEI condensed DNA, the PR_b functionalized stealth liposomes mediated better transfection than the non-targeted liposomes (Fig. 2). However, when the targeted formulations are compared, the bPEI condensed system performed over an order of magnitude better in terms of transfection efficiency than the pLL condensed system. These results are consistent with previous studies reported in the literature showing that bPEI condensed DNA performs better than the pLL counterpart in different gene delivery systems.15,16,19,75 Unencapsulated DNA particles condensed with either pLL or bPEI were both ineffective at transfecting DLD-1 cells to any appreciable extent under the same conditions. However, at higher concentrations of unencapsulated bPEI condensed DNA delivered, higher levels of transfection were seen (data not shown). At the same time non-targeted stealth liposomes encapsulating the same amount of DNA failed to give a noticeable level of transfection. These results demonstrate the stealth properties of these non-targeted liposomes, as the PEG brush layer is preventing these liposomes from interacting with and delivering their payload to cells. As our results show, transfection is successful when the PR_b targeting peptide is included on the stealth liposomes. bPEI–DNA free or encapsulated in PR_b functionalized stealth liposomes were non-toxic to cells (ESI, Fig. S2†).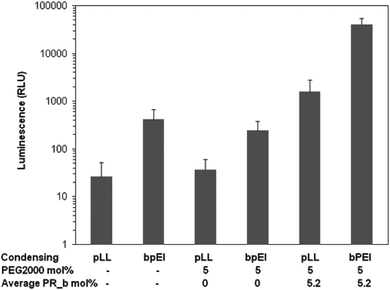 | ||
| Fig. 2 Transfection efficiency of liposome encapsulated and unencapsulated DNA condensed with bPEI or pLL in DLD-1 cells. 100 ng of DNA was delivered in different formulations to cells and incubated for 40 hours at 37 °C, after which luminescence was measured. The values represent mean ± standard error of two separate experiments (n = 2), each done in replicates of six. | ||
3.3 Effect of PR_b concentration on transfection
Having demonstrated the necessity of the PR_b peptide on our gene delivery vehicle, the PR_b concentration was optimized on stealth liposomes. As the concentration of the ligand is increased, an increase in the expression of luciferase in DLD-1 cells was observed (Fig. 3). At an average concentration of 1.5 mol% PR_b the transfection level of the targeted liposomes was only slightly higher than the non-targeted system. However at an average of 3 mol% a substantial difference was observed between transfection levels of non-targeted and targeted liposomes.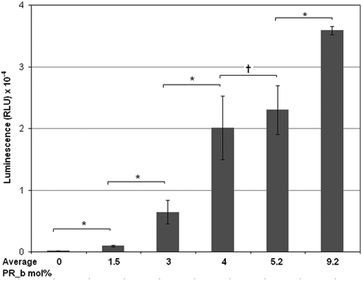 | ||
| Fig. 3 Effect of PR_b concentration on transfection efficiency of different 5 mol% PEG2000 stealth liposomal formulations encapsulating bPEI condensed DNA. 100 ng of DNA was delivered to DLD-1 cells in different formulations and incubated for 40 hours at 37 °C, followed by luminescence measurement. The values represent mean ± standard error of three separate liposomal experiments (n = 3), each done in replicates of six. Students t-test statistical analysis was performed and the statistical significance notated for the bracketed data * indicates p < 0.005; † indicates p > 0.5 and no statistical significance. | ||
Average concentrations of 4 and 5 mol% PR_b showed similar levels of transfection with no statistical difference between them, and both substantially higher than non-targeted liposomes. This increasing trend was maintained up to about 9 mol% PR_b, the highest concentration that we investigated. A similar trend with the PR_b concentration was seen in a previous study by our group delivering liposomes encapsulating a fluorescent dye to colorectal cancer cells,65 where conventional liposomes functionalized with PR_b between 0.7 and 3.5 mol% and PEGylated liposomes with PR_b between 1.1 and 2.6 mol% were tested. One hypothesis that can explain the effect of ligand concentration is the requirement of a certain minimum number of receptor–ligand interactions for the delivery vehicle to remain at the cell surface long enough to be internalized through a receptor mediated pathway. This requirement further adds to the specificity of the delivery vehicle. Healthy tissues which may have a lower number of integrin receptors may not achieve the required number of integrin–ligand interactions and therefore may not be transfected. Although the highest levels of transfection were obtained with 9 mol% PR_b, sufficiently high transfection levels were also observed at 4 and 5 mol% PR_b. Thus, for subsequent in vitro and in vivo work, liposomes were prepared with an average of 4 or 5 mol% PR_b concentration.
We did not examine peptide concentrations higher than 9 mol%, as previous work in our group demonstrated that Langmuir–Blodgett bilayers of peptide amphiphiles mixed with PEGylated lipids remained well-mixed at molar concentrations less than 10 mol% and higher than 35 mol%, while between 10 and 35 mol% the peptide amphiphiles phase separated.76
3.4 Demonstrating specificity of targeting
The GRGDSP peptide is a known binding ligand for integrins, including the α5β1 integrin. Blocking the integrin sites with an excess of free GRGDSP peptide would theoretically prevent the PR_b peptide from binding and thus hinder transfection. A 1000-fold excess of GRGDSP over the number of PR_b ligands on liposomes was used in these experiments, an average of 4 μg peptide per well in a 96 well plate. Additional GRGDSP was added every 8 hours to maintain an excess of blocking peptide, as this schedule proved to be more effective at blocking integrin binding compared to a large initial dose (data not shown). Fig. 4A shows that PR_b liposomes transfected unblocked cells at a level over an order of magnitude higher than the blocked liposomes. This experiment demonstrates the necessity of the integrin–PR_b interaction for successful transfection using the PR_b functionalized liposomal gene delivery vehicle. The results of these experiments are in agreement with our previously reported work where blocking experiments with peptides and antibodies demonstrated that PR_b is a specific ligand for the α5β1 integrin,65,69 with a dissociation constant of 76.3 ± 6.3 nM.77 To further investigate the specificity of the PR_b sequence for α5β1 integrin, a scrambled PR_b sequence (ScrPR_b) was designed as a negative control.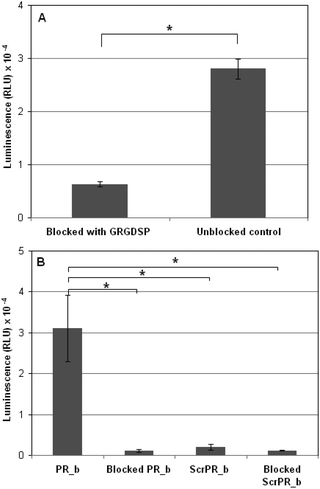 | ||
| Fig. 4 (A) Delivery of bPEI condensed DNA encapsulated in PR_b functionalized PEGylated liposomes to either unblocked DLD-1 cells or DLD-1 cells with their surface integrins blocked by GRGDSP peptides free in solution. Cells were incubated with 100 ng of DNA loaded stealth liposomes functionalized with 5 mol% PEG2000 and 5.3, 4 and 4 mol% PR_b (for the three liposomal formulations tested) for 40 hours at 37 °C after which the luminescence was quantified. (B) Delivery of bPEI condensed DNA encapsulated in PR_b functionalized or Scrambled PR_b functionalized PEGylated liposomes to either unblocked DLD-1 cells or DLD-1 cells with their surface integrins blocked by PR_b peptides free in solution. Cells were incubated with 100 ng of DNA loaded stealth liposomes functionalized with 5 mol% PEG2000 and 4.8, 6.1 and 5.9 mol% PR_b or 6.4, 5.4 and 5.9 mol% ScrPR_b for 40 hours at 37 °C after which the luminescence was quantified. The values represent mean ± standard error of three separate liposomal experiments (n = 3), each done in replicates of six. Students t-test statistical analysis was performed and the statistical significance notated for the bracketed data * indicates p < 0.001. | ||
Fig. 4B shows that in DLD-1 cells transfected with bPEI-DNA encapsulated in ScrPR_b functionalized stealth liposomes there was minimal luciferase expression when compared to that mediated by PR_b functionalized stealth liposomes. Furthermore, blocking with the free PR_b peptide inhibited the interaction of PR_b functionalized liposomes with DLD-1 cells almost completely. These results, taken together with the GRGDSP-blocking experiments shown in Fig. 4A, demonstrate the need for a specific integrin–PR_b interaction for efficient transfection.
3.5 In vivo gene delivery
Non-targeted stealth liposomes injected into tumor bearing mice (Fig. 5A) and PR_b functionalized stealth liposomes injected into healthy mice (Fig. 5B) showed minimal to no bioluminescent signal after one day. Considering that non-specific delivery to healthy organs continues to be a drawback for in vivo delivery vehicles,78 the lack of luciferase expression in the healthy mice may indicate minimum levels of gene delivery to healthy tissues using the PR_b functionalized stealth liposomes. When these liposomes were administered to tumor bearing mice, luciferase expression was seen preferentially in the region of the tumor, i.e., the liver (Fig. 5C). This metastatic tumor model has been designed to localize the tumor in the liver, and Fig. 6(A, B) is an ex vivo image of the dissected abdomen from mouse 12 in Fig. 5C, clearly showing the presence of luciferase expressing tumor localized in the liver.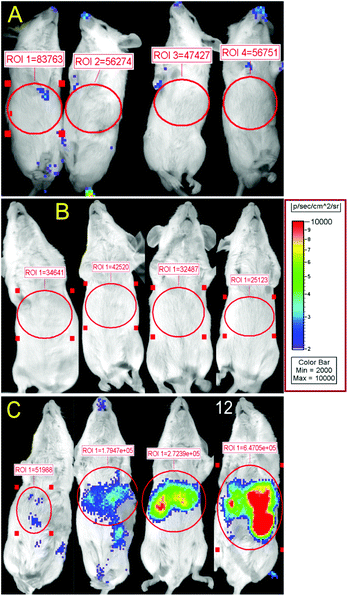 | ||
| Fig. 5 Bioluminescence from luciferase expression after administration of 700 ng of bPEI-condensed DNA encapsulated in different stealth liposome formulations. 24 hours after liposome injection expression of luciferase was visualized by bioluminescent imaging. Total relative optical intensity (ROI) is shown for each image as measured in photons/second/cm2 by the Xenogen IVIS Imaging System. (A) Non-targeted stealth liposomes, with 5 mol% PEG2000, delivered to CT26 tumor bearing mice. (B) 3.3 mol% PR_b functionalized stealth liposomes (5 mol% PEG2000) delivered to healthy mice. (C) 3.3 mol% PR_b and 5 mol% PEG2000 targeted stealth liposomes delivered to CT26 tumor bearing mice. | ||
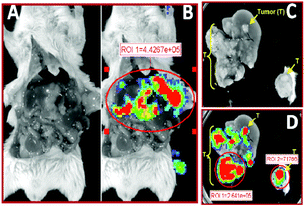 | ||
| Fig. 6 Ex vivo bioluminescence from luciferase expression after administration of 3.3 mol% PR_b, 5 mol% PEG2000 functionalized stealth liposomes in the CT26 tumor bearing mouse 12 shown in Fig. 5. Total relative optical intensity (ROI) as measured in photons/second/cm2 by the Xenogen IVIS Imaging System is shown for each image lateral dissection showing tumor load on liver (A) with superimposed bioluminescence (B). Excised liver showing white tumor nodules (C) with superimposed bioluminescence (D). | ||
Our preliminary results are in contrast to previous studies targeting the integrins in vivo in which significant non-specific uptake in the liver was reported.46,48–50Ex vivo images of the liver are shown in Fig. 6C, and a closer inspection shows bioluminescence in the liver tissue originating from the tumor nodules, and minimal signal from the surrounding healthy liver tissue (Fig. 6D). The essence of targeted therapy is in the specificity of delivery – in the ability to differentiate between healthy organs and the diseased site. Fig. 5 and 6 show that PR_b functionalized stealth liposomes may have the potential to differentiate not only between healthy organs and tumor bearing organs, but also between tumor tissue and healthy tissue within the same organ. Our preliminary in vivo studies show that the PR_b functionalized stealth liposome warrants further investigation as an in vivo delivery vehicle for therapeutic payloads.
In addition, most in vivo studies report administrations of anywhere from 5 to 50 μg of DNA per mouse.6,70,79,80 A high dose combined with poor specificity can result in significant undesirable accumulation of therapeutics in healthy tissues. In this paper we have demonstrated the in vitro specificity of the PR_b functionalized liposome, and shown in our in vivo work that we can potentially achieve gene expression specifically in the target site with less than 1 μg DNA injected per mouse. However, whether this expression level is enough to achieve a therapeutic effect still needs to be investigated.
4. Conclusion
We have designed and developed a gene delivery vehicle that encapsulates bPEI condensed DNA in PR_b functionalized stealth liposomes. The PR_b targeted vehicle is biocompatible, non toxic, stable to aggregation, and specifically targets cells overexpressing α5β1 integrin with the potential to differentiate between healthy and diseased tissue within the same organ. Many diseased cells including cancers often overexpress α5β1 integrin, making the PR_b functionalized stealth liposome a potentially effective therapeutic delivery agent that may specifically reach the diseased site while sparing healthy tissues.Acknowledgements
We thank Kelly Podetz-Pederson for technical assistance. The cryo-TEM experiments included in this work were carried out in the University of Minnesota Characterization Facility, which receives partial support from NSF through the NNIN program. Graduate Research Fellowships are acknowledged from the National Science Foundation for RML and the National Institute of Dental and Craniofacial Research (F30DE020210) for BSS. This work was supported by the National Cancer Institute (R01CA120383). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute.Notes and references
- R. Niven, R. Pearlman, T. Wedeking, J. Mackeigan, P. Noker, L. Simpson-Herren and J. G. Smith, J. Pharm. Sci., 1998, 87, 1292–1299 CrossRef CAS.
- R. Kircheis, S. Schuller, S. Brunner, M. Ogris, K. H. Heider, W. Zauner and E. Wagner, J. Gene Med., 1999, 1, 111–120 CrossRef CAS.
- X. B. B. Zhao and R. J. Lee, Adv. Drug Delivery Rev., 2004, 56, 1193–1204 CrossRef CAS.
- L. Wang, W. Su, Z. Liu, M. Zhou, S. Chen, Y. Chen, D. Lu, Y. Liu, Y. Fan, Y. Zheng, Z. Han, D. Kong, J. C. Wu, R. Xiang and Z. Li, Biomaterials, 2012, 33, 5107–5114 CrossRef CAS.
- K. Kunath, A. von Harpe, D. Fischer, H. Peterson, U. Bickel, K. Voigt and T. Kissel, J. Controlled Release, 2003, 89, 113–125 CrossRef CAS.
- C. Fu, L. Lin, H. Shi, D. Zheng, W. Wang, S. Gao, Y. Zhao, H. Tian, X. Zhu and X. Chen, Biomaterials, 2012, 33, 4589–4596 CrossRef CAS.
- Y. I. Kim, B. C. Ahn, J. A. Ronald, R. Katzenberg, A. Singh, R. Paulmurugan, S. Ray, S. S. Gambhir and L. V. Hofmann, J. Vasc. Intervent. Radiol., 2012, 23, 704–711 CrossRef.
- C. Cole, J. Qiao, T. Kottke, R. M. Diaz, A. Ahmed, L. Sanchez-Perez, G. Brunn, J. Thompson, J. Chester and R. G. Vile, Nat. Med., 2005, 11, 1073–1081 CrossRef CAS.
- K. L. Douglas, Biotechnol. Prog., 2008, 24, 871–883 CAS.
- S.-D. Li and L. Huang, J. Controlled Release, 2007, 123, 181–183 CrossRef CAS.
- M. A. Mintzer and E. E. Simanek, Chem. Rev., 2009, 109, 259–302 CrossRef CAS.
- M. Chaszczewska-Markowska, M. Ugorski and M. Langner, Cell. Mol. Biol. Lett., 2004, 9, 3–13 CAS.
- I. Moret, J. E. Peris, V. M. Guillem, M. Benet, F. Revert, F. Dasi, A. Crespo and S. F. Alino, J. Controlled Release, 2001, 76, 169–181 CrossRef CAS.
- D. D. Stuart, G. Y. Kao and T. M. Allen, Cancer Gene Ther., 2000, 7, 466–475 CAS.
- C. Y. M. Hsu and H. Uludag, BMC Biotechnol., 2008, 8 Search PubMed.
- M. Mannisto, M. Reinisalo, M. Ruponen, P. Honkakoski, M. Tammi and A. Urtti, J. Gene Med., 2007, 9, 479–487 CrossRef CAS.
- Y. Yue, F. Jin, R. Deng, J. Cai, Z. Dai, M. C. M. Lin, H.-F. Kung, M. A. Mattebjerg, T. L. Andresen and C. Wu, J. Controlled Release, 2011, 152, 143–151 CrossRef CAS.
- J. P. Behr, Chimia, 1997, 51, 34–36 CAS.
- O. Boussif, F. Lezoualch, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297–7301 CrossRef CAS.
- A. Akinc, M. Thomas, A. M. Klibanov and R. Langer, J. Gene Med., 2005, 7, 657–663 CrossRef CAS.
- W. T. Godbey, K. K. Wu and A. G. Mikos, Biomaterials, 2001, 22, 471–480 CrossRef CAS.
- D. Chen, Y. Ping, G. Tang and J. Li, Soft Matter, 2010, 6, 955–964 RSC.
- L. Chen, H. Tian, J. Chen, X. Chen, Y. Huang and X. Jing, J. Gene Med., 2010, 12, 64–76 CrossRef CAS.
- Y.-X. Sun, W. Xiao, S.-X. Cheng, X.-Z. Zhang and R.-X. Zhuo, J. Controlled Release, 2008, 128, 171–178 CrossRef CAS.
- C.-F. Wang, Y.-X. Lin, T. Jiang, F. He and R.-X. Zhuo, Biomaterials, 2009, 30, 4824–4832 CrossRef CAS.
- M. S. Lee, M. G. Kim, Y. L. Jang, K. Lee, T. G. Kim, S. H. Kim, T. G. Park, H. T. Kim and J. H. Jeong, Macromol. Res., 2011, 19, 688–693 CrossRef CAS.
- Y. H. Kim, J. H. Park, M. Lee, T. G. Park and S. W. Kim, J. Controlled Release, 2005, 103, 209–219 CrossRef CAS.
- R. Deng, Y. Yue, F. Jin, Y. Chen, H.-F. Kung, M. C. M. Lin and C. Wu, J. Controlled Release, 2009, 140, 40–46 CrossRef CAS.
- H. Cheng, J.-L. Zhu, X. Zeng, Y. Jing, X.-Z. Zhang and R.-X. Zhuo, Bioconjugate Chem., 2009, 20, 481–487 CrossRef CAS.
- Y. Nie, D. Schaffert, W. Roedl, M. Ogris, E. Wagner and M. Guenther, J. Controlled Release, 2011, 152, 127–134 CrossRef CAS.
- S. Yamano, J. Dai, C. Yuvienco, S. Khapli, A. M. Moursi and J. K. Montclare, J. Controlled Release, 2011, 152, 278–285 CrossRef CAS.
- Y. T. Ko, R. Bhattacharya and U. Bickel, J. Controlled Release, 2009, 133, 230–237 CrossRef CAS.
- L. Garcia, M. Bunuales, N. Duzgunes and C. T. de Ilarduya, Eur. J. Pharm. Biopharm., 2007, 67, 58–66 CrossRef CAS.
- C. H. Lee, Y. H. Ni, C. C. Chen, C. K. Chou and F. H. Chang, Biochim. Biophys. Acta, Biomembr., 2003, 1611, 55–62 CrossRef CAS.
- J. Pelisek, L. Gaedtkel, J. DeRouchey, G. F. Walkerl, S. Niko and E. Wagner, J. Gene Med., 2006, 8, 186–197 CrossRef CAS.
- J. Schafer, S. Hobel, U. Bakowsky and A. Aigner, Biomaterials, 2010, 31, 6892–6900 CrossRef.
- D. C. Drummond, O. Meyer, K. L. Hong, D. B. Kirpotin and D. Papahadjopoulos, Pharmacol. Rev., 1999, 51, 691–743 CAS.
- D. X. Liu, A. Mori and L. Huang, Biochim. Biophys. Acta, 1992, 1104, 95–101 CrossRef CAS.
- V. P. Torchilin, V. G. Omelyanenko, M. I. Papisov, A. A. Bogdanov, V. S. Trubetskoy, J. N. Herron and C. A. Gentry, Biochim. Biophys. Acta, Biomembr., 1994, 1195, 11–20 CrossRef CAS.
- M. C. Woodle, K. K. Matthay, M. S. Newman, J. E. Hidayat, L. R. Collins, C. Redemann, F. J. Martin and D. Papahadjopoulos, Biochim. Biophys. Acta, 1992, 1105, 193–200 CrossRef CAS.
- D. C. Litzinger, A. M. J. Buiting, N. Vanrooijen and L. Huang, Biochim. Biophys. Acta, Biomembr., 1994, 1190, 99–107 CrossRef CAS.
- P. L. Ahl, S. K. Bhatia, P. Meers, P. Roberts, R. Stevens, R. Dause, W. R. Perkins and A. S. Janoff, Biochim. Biophys. Acta, Biomembr., 1997, 1329, 370–382 CrossRef CAS.
- P. G. Waser, U. Muller, J. Kreuter, S. Berger, K. Munz, E. Kaiser and B. Pfluger, Int. J. Pharm., 1987, 39, 213–227 CrossRef CAS.
- I. Ahmad and T. M. Allen, Cancer Res., 1992, 52, 4817–4820 CAS.
- M. Liu, C. Li, M. Pazgier, C. Q. Li, Y. B. Mao, Y. F. Lv, B. Gu, G. Wei, W. R. Yuan, C. Y. Zhan and W. Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14321–14326 CrossRef CAS.
- R. M. Schiffelers, G. A. Koning, T. L. M. ten Hagen, M. Fens, A. J. Schraa, A. Janssen, R. J. Kok, G. Molema and G. Storm, J. Controlled Release, 2003, 91, 115–122 CrossRef CAS.
- T. O. Pangburn, M. A. Petersen, B. Waybrant, M. M. Adil and E. Kokkoli, J. Biomech. Eng., 2009, 131, 074005 CrossRef.
- J. Jiang, S.-j. Yang, J.-c. Wang, L.-j. Yang, Z.-z. Xu, T. Yang, X.-y. Liu and Q. Zhang, Eur. J. Pharm. Biopharm., 2010, 76, 170–178 CrossRef CAS.
- P. K. Dubey, V. Mishra, S. Jain, S. Mahor and S. P. Vyas, J. Drug Targeting, 2004, 12, 257–264 CrossRef CAS.
- M. H. A. M. Fens, K. J. Hill, J. Issa, S. E. Ashton, F. R. Westwood, D. C. Blakey, G. Strom, A. J. Ryan and R. M. Schiffelers, Br. J. Cancer, 2008, 99, 1256–1264 CrossRef CAS.
- J. Gong, D. Wang, L. Sun, E. Zborowska, J. K. V. Willson and M. G. Brattain, Cell Growth Differ., 1997, 8, 83–90 CAS.
- D. G. Jayne, R. M. Heath, O. Dewhurst, N. Scott and P. J. Guillou, Eur. J. Surg. Oncol., 2002, 28, 30–36 CrossRef CAS.
- Y. F. Jia, Z. Z. Zeng, S. M. Markwart, K. F. Rockwood, K. M. W. Ignatoski, S. P. Ethier and D. L. Livant, Cancer Res., 2004, 64, 8674–8681 CrossRef CAS.
- C. M. Witkowski, I. Rabinovitz, R. B. Nagle, K. S. D. Affinito and A. E. Cress, J. Cancer Res. Clin. Oncol., 1993, 119, 637–644 CrossRef CAS.
- S. Kim, K. Bell, S. A. Mousa and J. A. Varner, Am. J. Pathol., 2000, 156, 1345–1362 CrossRef CAS.
- A. Maglott, P. Bartik, S. Cosgun, P. Klotz, P. Ronde, G. Fuhrmann, K. Takeda, S. Martin and M. Dontenwill, Cancer Res., 2006, 66, 6002–6007 CrossRef CAS.
- A.-M. C. Dingemans, V. van den Boogaart, B. A. Vosse, R.-J. van Suylen, A. W. Griffioen and V. L. Thijssen, Mol. Cancer, 2010, 9 Search PubMed.
- J. L. Muschler and A. F. Horwitz, Development, 1991, 113, 327–337 CAS.
- C. H. Chen, D. Z. Liu, H. W. Fang, H. J. Liang, T. S. Yang and S. Y. Lin, Biosci. Biotechnol. Biochem., 2008, 72, 1586–1594 CrossRef CAS.
- C. B. Pattillo, F. Sari-Sarraf, R. Nallamothu, B. M. Moore, G. C. Wood and M. F. Kiani, Pharm. Res., 2005, 22, 1117–1120 CrossRef CAS.
- Y.-f. Zhang, J.-c. Wang, D.-y. Bian, X. Zhang and Q. Zhang, Eur. J. Pharm. Biopharm., 2010, 74, 467–473 CrossRef CAS.
- H. Zhao, J.-C. Wang, Q.-S. Sun, C.-L. Luo and Q. Zhang, J. Drug Targeting, 2009, 17, 10–18 CrossRef CAS.
- A. Mardilovich, J. A. Craig, M. Q. McCammon, A. Garg and E. Kokkoli, Langmuir, 2006, 22, 3259–3264 CrossRef CAS.
- J. A. Craig, E. L. Rexeisen, A. Mardilovich, K. Shroff and E. Kokkoli, Langmuir, 2008, 24, 10282–10292 CrossRef CAS.
- A. Garg, A. W. Tisdale, E. Haidari and E. Kokkoli, Int. J. Pharm., 2009, 366, 201–210 CrossRef CAS.
- D. Demirgöz, A. Garg and E. Kokkoli, Langmuir, 2008, 24, 13518–13524 CrossRef.
- A. Garg and E. Kokkoli, Curr. Pharm. Biotech., 2011, 12, 1135–1143 CrossRef CAS.
- N. A. Atchison, W. Fan, K. K. Papas, B. J. Hering, M. Tsapatsis and E. Kokkoli, Langmuir, 2010, 26, 14081–14088 CrossRef CAS.
- T. O. Pangburn, F. S. Bates and E. Kokkoli, Soft Matter, 2012, 8, 4449–4461 RSC.
- L. R. Belur, R. S. McIvor and A. Wilber, Methods Mol. Biol., 2008, 434, 267–276 CAS.
- B. Lucas, K. Remaut, N. N. Sanders, K. Braeckmans, S. C. De Smedt and J. Demeester, Biochemistry, 2005, 44, 9905–9912 CrossRef CAS.
- P. Berndt, G. B. Fields and M. Tirrell, J. Am. Chem. Soc., 1995, 117, 9515–9522 CrossRef CAS.
- F. Szoka and D. Papahadjopoulos, Annu. Rev. Biophys. Bioeng., 1980, 9, 467–508 CrossRef CAS.
- C. H. Fiske and Y. Subbarow, J. Biol. Chem., 1925, 66, 375–400 CAS.
- Tarwadi, J. A. Jazayeri, R. J. Prankerd and C. W. Pouton, Bioconjugate Chem., 2008, 19, 940–950 CrossRef CAS.
- A. Mardilovich and E. Kokkoli, Langmuir, 2005, 21, 7468–7475 CrossRef CAS.
- K. Shroff, T. R. Pearce and E. Kokkoli, Langmuir, 2012, 28, 1858–1865 CrossRef CAS.
- T. R. Pearce, K. Shroff and E. Kokkoli, Adv. Mater., 2012, 24, 3803–3822 CrossRef CAS.
- L. R. Belur, K. M. Podetz-Pedersen, B. S. Sorenson, A. H. Hsu, J. B. Parker, C. S. Carlson, D. A. Saltzman, S. Ramakrishnan and R. S. McIvor, Mol. Cancer, 2011, 10, 12 CrossRef.
- C. Gong, X. Li, L. Xu and Y.-H. Zhang, Biomaterials, 2012, 33, 3456–3463 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1, Size and zeta potential measurements of pLL and bPEI condensed DNA particles; Fig. S2, Cell viability after treatment with transfection agents. See DOI: 10.1039/c2bm00128d |
| This journal is © The Royal Society of Chemistry 2013 |