Taking tissue adhesives to the future: from traditional synthetic to new biomimetic approaches
Lígia Pereira
Bré
ab,
Yu
Zheng
b,
Ana Paula
Pêgo
*ac and
Wenxin
Wang
*b
aFaculdade de Engenharia da Universidade do Porto, Rua Dr. Roberto Frias, s/n 4200-465, Porto, Portugal. E-mail: feup@fe.up.pt; Fax: +351 22 508 1440; Tel: +351 22 508 14 00
bNetwork of Excellence for Functional Biomaterials (NFB), National University of Ireland, Galwal, IDA Business Park, Dangan, Galway, Ireland. E-mail: wenxin.wang@nuigalway.ie; Fax: +353 91 495 585; Tel: +353 91 49 5833
cInstituto de Engenharia Biomédica (INEB), Rua do Campo Alegre, 4150-180, Porto, Portugal. E-mail: apego@ineb.up.pt; Fax: +351 226094567; Tel: +351 226074900
First published on 26th November 2012
Abstract
Tissue adhesives are a versatile and valuable alternative for wound closure. With fast application, the prevention of body fluid leakage and additional trauma to the wound, adhesives are desirable, especially in friable tissues. Although several options are already in the market, these present some drawbacks, namely poor adhesion in wet substrates and toxicity. Here, the main adhesives both synthetic and biomimetic, commercially available and those still in research, are analyzed with a focus on their adhesion mechanisms. At present, the strongest adhesive able to replace sutures in skin wounds is cyanoacrylate based. However it is toxic and therefore cannot be used internally. More recently alternatives have emerged, with PEG-based adhesives being biocompatible but mainly used as tissue sealants due to their low strength. The most recent approaches under development try to mimic the adhesion strategies of several organisms, such as the extensively studied blue mussels, sandcastle worms, barnacles and geckos. Although no ideal results have been achieved so far, large improvements have been accomplished in the last decade and research is reaching to a new level where the results are starting to meet the needs for medical applications.
 Lígia Pereira Bré | Lígia Bré was born in Porto, Portugal in 1989. She received her B.Sc. (2010) and M.Sc. (2012) degrees in Bioengineering, speciality in Biomedical Engineering from the Faculty of Engineering of the University of Porto. She first visited the Network of Excellence for Functional Biomaterials in Galway, Ireland in 2011 as an exchange student. In 2012 she was invited to return to the lab for her Masters thesis where she worked on biomimetic tissue adhesives. |
 Yu Zheng | Yu Zheng was born in Wuhan, China in 1983. He received his M.Sc. in Polymer Science from University of Sheffield, where he worked on asymmetrical polymer crystallization structure and growth rate. His Ph.D. degree was obtained from University of Nottingham in the synthesis of dendritic polymers in 2010. He is currently a postdoctoral fellow at the Network of Excellence for Functional Biomaterials (NFB) in National University of Ireland, Galway, working in the group of Dr Wenxin Wang. |
 Ana Paula Pêgo | Ana Pêgo (Ph.D. in Polymer Chemistry and Biomaterials, University of Twente, Netherlands) is currently a Principal Investigator at INEB—Instituto de Engenharia Biomédica (Portugal) and Invited Professor at the University of Porto. She is co-coordinator of the NEWTherapies Group and leader of the Biomaterials for Neurosciences team. Her research on new biomaterials for application in neurosciences includes the development of new polymers for the design of biomaterial-based vectors for gene therapy and the preparation of nerve grafts for spinal cord injury treatment. Recently she has been appointed the Scientific Director of the Bioimaging Centre for Biomaterials and Regenerative Therapies of INEB. |
 Wenxin Wang | Dr Wenxin Wang received his bachelor's degree from Si Chuan University. He obtained his M.Sc. and Ph.D. in polymer synthesis from the Shanghai Jiao Tong University in 1996 and 1999, respectively. After his Ph.D., he worked as a research fellow at University of Liege (Belgium) in 2000 and University of Nottingham (UK) from 2001–2008. In 2008, he was appointed as University fellow at the National University of Ireland Galway and soon promoted to Stokes Lecturer in Biomedical Engineering. Dr Wenxin Wang leads a team of 12 researchers working at the interface of polymer and life science. |
Introduction
Surgical sutures, staples and clips have been used throughout the world in the context of wound closure and tissue reconstruction. The use of some goes back to the ancient history and little evolution has been seen.1 Although their effectiveness has been proven, in many circumstances the existing products are unable to provide efficient wound closure, and in some situations cannot be used at all as they interfere with the functional rehabilitation of the tissue.2 In some cases, suturing friable tissues such as the dura mater,3 lung,4 liver,5 spleen and kidney6 is not a suitable solution and this is also true in stopping the leakage of body fluids. Even when traditional solutions are applied they still can cause additional trauma to the wound and need follow up, which raises the costs of the wound management procedure.In order to improve wound healing and to overcome these obstacles, adhesives appear to be an ideal alternative solution. They can be easily applied on top of the wound, act as blood and humidity sealants, create an antimicrobial barrier, cause less trauma and result in a satisfactory cosmetic outcome.7,8
Considering that every year in the United States of America, more than 12 million traumatic wounds are treated in the Emergency Departments and that an average cost of $21.58 per wound has been reported, more than $258 million is spent on suturing kits alone.9 Even though the cost per unit of some adhesives is higher than that of sutures, a study that accounted for all the equipment, health care workers’ time and follow up visits proved the adhesive's cost effectiveness.10 Despite this fact, 60% of wound closure technologies are still represented by sutures, staples and other mechanical methods.11 However, hemostats, sealants and adhesives have seized a portion of this market and recent studies reveal an estimated future global market potential of $38 billion for adhesive by 2017.12
This promising market has been the driving force for the development of new strategies, as the currently available commercial tissue adhesives still demand improvement. With current adhesives, when optimal strength is achieved, toxicity is also present2 and when these are applied in wet conditions adhesion is normally compromised.13 Inspired by nature and combining the knowledge of its multiple adhesive mechanisms with science and technology, researchers have unveiled different biomimetic adhesives. From the bioadhesive groups of blue mussels14 to the micropatterned surfaces to mimic gecko's feet,15 many options are available.
With all the research that has been done over the last decade, a number of reviews have described the different types of materials (natural and synthetic) for tissue adhesives.2,16 However, biomimetic approaches are still a very recent reality in which advances are made every year. This review will focus on the work developed in the last ten years with special emphasis on the mechanisms responsible for the adhesive's performance, both in traditional synthetic and newly biomimetic inspired approaches including mussels, sandcastle worms and geckos.
Synthetic/semi-synthetic adhesives
For many decades, as natural-based alternatives for wound closure were scarce and existing ones could not be used for a variety of reasons including high costs or low bonding strength, others have been explored. Synthetic adhesives, where the most popular ones are cyanoacrylates, have two very useful advantages: high bonding strength—the highest known in the market—and low cost of production. The control of physical and chemical properties is also a considerable advantage in such adhesives. However, there is a major disadvantage that makes them available for internal use in only a few cases. Degradation products accumulate in tissue and are toxic.17 In order to overcome this difficulty, improvements in cyanoacrylates to reduce toxicity have been made as well as research carried out using biocompatible polymers. In this section, the mechanism of cyanoacrylate adhesion and its applications will be discussed as well as strategies using functionalized polyethylene glycol, castor oil and urethane adhesives (Table 1).| Composition | Adhesion test | Strength (kPa) | Crosslinking | Application | Side effects | References |
|---|---|---|---|---|---|---|
| Cyanoacrylates | Tensile strength | ∼68 | Water | Skin | Cytotoxic | 2,16,18,19 |
| Cornea | ||||||
| Gastric therapy | ||||||
| Laparoscopic surgery | ||||||
| PEG | Burst pressure and tensile strength | 13 | Chemical or light | Dura mater | Possible crosslinker toxicity | 2,20,21 |
| Cardiovascular surgery | ||||||
| Liver | ||||||
| CS-PEG | Tensile strength | 50 | Blue light | Cartilage | n.a. | 22 |
| Dextran | Tensile strength | ∼300 | UV light | n.a. | UV damage | 23 |
| Urethane | Tensile strength | n.a. | Chemical or UV radiation | Bone | Thrombogenecity | 24,25 |
Cyanoacrylates
Cyanoacrylates first appeared in 1942, developed for industrial purposes. They are a class of synthetic glues composed of monomers (esters of cyanoacrylic acid) which polymerize in seconds upon contact with weak bases such as water, blood or cell membranes26 (mechanism shown in Fig. 1). An exothermic reaction occurs during the polymerization process, creating a strong yet flexible film which bonds the apposed wound edges.27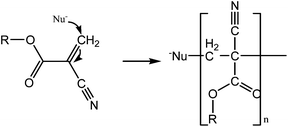 | ||
| Fig. 1 Mechanism of general cyanoacrylate polymerization in contact with a nucleophile. The C–C double bond is highly reactive and the chain is formed in seconds. | ||
There are several advantages from this mechanism, among which are strong bonding properties on a dry operation field,2 the fast setting reaction28 and a relatively low cost. The adhesive can also function as a microbial barrier.29 In addition it is also important to state the ability to tune the bonding properties through modifying the alkyl side chains in the cyanoacrylic acid structure. This one in fact becomes very important as the toxicity—the major obstacle to its use in medical applications—can be reduced as the carbon number in the ester side chains is increased.30 For instance, with 10 carbons in the side chain, the compound becomes less toxic whereas if the carbon number is less than 4, the glue degrades rapidly, with degradation products (cyanoacetate and formaldehyde) accumulated in the tissue that can cause both acute and chronic inflammation.2,19
The major concerns with toxicity, as well as with mechanical properties, led to a series of improvements after the release of the first cyanoacrylate-based medical adhesives on the market. Various homologues (Fig. 2) were developed and newer properties and needs have been directing research and clinical use. N-Butyl-2-cyanoacrylate has been widely used in endoscopic therapy for more than 10 years, replacing the initially applied butyl-cyanoacrylates which have reported inferior mechanical properties. Similarly, in 1998, the introduction of 2-octyl-cyanoacrylate (OCA), with higher tensile strength, was a landmark in wound healing research. This adhesive is still used in skin closure and can be successfully applied on surgical incisions from 0.1 cm to more than 50 cm long.31 With an average strength of 68 kPa it is indicated for cornea, skin wounds or endoscopic therapy.2,16
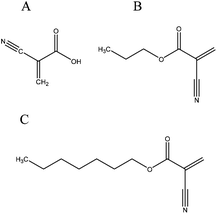 | ||
| Fig. 2 Structure of (A) cyanoacrylic acid, (B) n-butyl-cyanoacrylate and (C) octyl-cyanoacrylate. | ||
Quite a few products are commercially available, differing both in formulation and in applicator design. The most famous n-butyl-cyanoacrylates are Histoacryl (Braun, Melsungen, Germany)32 whose application is described in more than a 1200 publications and Vetbond™ (3M, St. Paul, Minnesota, US).33 Glubran2 (GEM s.r.l., Viareggio, Italy) appears as a modified n-butyl-2-cyanoacrylate glue approved for internal and external use in Europe and it is mainly used for endoscopic therapy.34 LiquiBand Surgical® (MedLogic Global Limited, Plymouth, UK) is an octyl-blend formulation that combines the fast setting properties of butyl-cyanoacrylates with a more flexible liquid wound dressing capability provided by the slower setting octyl-cyanoacrylates.35 The most popular glue for skin closure nowadays is Dermabond® (Ethicon, Inc., Somerville, NJ), a 2-octyl-cyanoacrylate which has been prospectively compared with sutures.36 This adhesive has been rated less painful by patients undergoing repair of cutaneous lacerations and studies report a decreased time of repair when using it.37
There is still one major difficulty regarding cyanoacrylate adhesives. Despite all the efforts to reduce toxicity, it cannot be used for internal applications. In fact, even in skin wounds, it is important to prevent contact between the glue and the wound itself because the glue would cause histotoxic reactions if trapped under apposed skin edges. In general, four main types of complications may occur due to the application of cyanoacrylates: systemic inflammatory reaction; local tissue necrosis; thrombo-embolic complications or septic complications.16
Poly ethylene glycol based adhesives
As toxicity is one of the major concerns in adhesives, there is the need to design solutions to overcome that obstacle. Polyethylene glycol (PEG) is a compound with numerous applications from industry to medicine. PEG polymers are biodegradable and can be normally excreted by the human body. The adhesion mechanism in PEG-based systems is highly dependent on the formulation used, presenting high variability due to the readily modifiable chemistries.38 This variability led many groups to use different PEG-based hydrogels for tissue adhesion.FocalSeal™ (Focal, Lexington, MA) is an adhesive designed to seal air leaks associated with lung and cardiac surgery20 and cerebral spinal fluid (CSF) leakage after neurosurgical dural repair.39 It is a two compound system: an eosin primer which is first applied and the solution comprised of PEG which is applied after the first element undergoes self-crosslinking. It also needs to be activated by light and requires a significant amount of time for the procedure which makes it unsuitable for hemorrhagic situations. Chronic inflammation responses with macrophages and giant cells are reported complications associated with its use.2 CoSeal® (Baxter Healthcare International, Palo Alto, CA) is another commercial product but, unlike FocalSeal™, its polymerization is chemically activated. When studying its mechanical properties using a biaxial tensile tester, Azadani et al. found that its stiffness is very similar to that of Tisseel®, a fibrin product of the same company. Furthermore, CoSeal® showed better sealing and haemostatic performance than Tisseel®.40 Currently it is being used for cardiovascular surgery.
Artzi et al. proposed an adhesive prepared with dextran aldehyde (Fig. 3) and an 8-arm PEG amine which crosslinks within minutes. The adhesion mechanism is ascribed to the interaction between aldehyde groups in the adhesive and amine groups in tissues. This allows the possibility of providing tissue specific adhesions through the control of aldehyde groups. A test using rat small intestine was performed to assess adhesive strength. Maximum strength was around 13.6 kPa and was bracketed by those of fibrin and cyanoacrylate.21
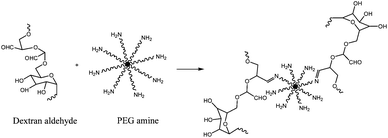 | ||
| Fig. 3 Reaction between dextran aldehyde and PEG amine formed an adhesive that crosslinks within minutes.21 | ||
Also using dextran but following a different approach, Li et al. described a photocrosslinkable adhesive composed of 2-isocyanatoethyl methacrylate (IEMA) chemically modified onto a dextran polymer backbone. A maximum strength at breakage of 299 kPa was achieved. With normal cell morphology and good distribution, the authors suggest good cell viability; however, statistical differences compared to the negative control were observed.23 These results again imply that although a certain degree of biocompatibility in tissue adhesives is being achieved, improvements are still needed in the majority of formulations.
A chondroitin sulfate PEG (CS-PEG) hydrogel material with covalent adhesion to tissue, again through amine bonds, was developed by Strehin et al. Adhesion was quantified using a lap shear test on bovine articular cartilage. It nearly reached 50 kPa and was 10 times stronger than the fibrin control. It was also biodegradable, via hydrolysis and enzymatically, and no toxicity was found.22
A different strategy is composed of 4-arm PEG maleimide (Mw = 10 kDa), 4-arm PEG thiol (Mw = 10 kDa) and silk fibroin. Silk PEG gel formation occurs in two phases. Phase I includes the chemical crosslinking between the two types of PEG (shown in Fig. 4), and in phase II the network is stabilized through silk β-sheet formation. Adhesive properties were comparable to those of CoSeal and decreased swelling and longer degradation times were observed.41 Other approaches have been developed using functionalization with bioadhesive moieties. Those shall be discussed further ahead.
 | ||
| Fig. 4 Phase I of adhesive formation: crosslinking scheme between the PEG maleimide and PEG thiol.41 | ||
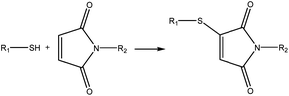
Urethane based adhesives
Recently, urethane based adhesives have also been considered for use as soft tissue adhesives. Possible biocompatibility and biodegradability, as well as the ability to form urea linkages through reaction with the amino groups present in the biological molecules of the tissue to promote adhesion (see Fig. 5) are the main properties that make these appealing. Still, research in this area is quite limited. Studies of poly urethanes as adhesives go back to the 80's.42 With no toxic effects, urethane-based sealants were tested and showed efficacy in renal surgery, endocrinology and pancreatic occlusion.43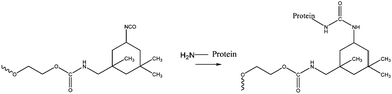 | ||
| Fig. 5 Model of urethane tissue adhesion through the reaction with the amino groups in the biological molecules. Formation of covalent bonds.43 | ||
Ferreira et al. proceeded to the modification of castor oil with free isocyanate groups to apply it as a bioadhesive. While haemocompatibility and thrombogenecity were evaluated, the mechanical tests performed are not comparable to the literature because only force is presented. Gelatin sheets were used as the substrate and no adhesion occurred in dry samples. Urethanes are cured by moisture rather than air so maximum force was achieved when the substrate was moisturized with water: 150 N.24 Later, the same group worked on the development of poly caprolactone-based (PCL) urethanes synthesized through the reaction with isophorone diisocyanate (IPD) or hexamethylene diisocyanate (HDI).44 Adhesion force was evaluated following the previous method (gelatin sheets) and different results were obtained for the two variations of the urethane adhesives. PCL-IPD breakage occurred in the gelatin piece showing high adhesion force and PCL-HDI failed adhesion. Performed tests assess the adhesive as a thrombogenic material which is related to its low surface energy but the authors consider this an advantage pointing to its haemostatic character as a valuable property in tissue adhesion.
Despite other studies proving its adhesion, the curing time of urethane adhesives is still not suitable for surgical procedures on soft tissues.43 In order to overcome this disadvantage, PCL was modified with 2-isocyanate ethylmethacrylate to form a photocrosslinkable adhesive via UV irradiation.45
With both advantages and disadvantages, urethane adhesion led to the development of a bone adhesive, Kryptonite™ Bone Cement (Doctors Research Group).25 Approved by the FDA (Food and Drug Administration) since 2009, the adhesive which is 5 times stronger than PMMA (poly(methylmethacrylate glue) consists of three components. The first, the pre-polymer, is a hydroxyl-terminated fatty acid derived from castor oil, synthesized with NCO groups. The second, a polyol, also contains the hydroxyl-terminated fatty acid, along with water and a catalyst. The water and NCO groups react and release CO2 (Fig. 6) which is one of the sources of the adhesive's porosity along with the third component, calcium carbonate. The last provides the structural and mechanical properties and allows the adhesive to act as a scaffold for bone regeneration.
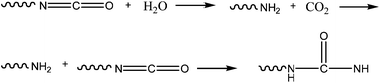 | ||
| Fig. 6 Kryptonite adhesive polymerization mechanism. | ||
The functional groups of the two first components are available to chemically bond with both organic (bone) and inorganic (stainless steel) surfaces and the expansion caused by polymerization introduces macro adhesion. Viscosity varies with time from an injectable liquid in the first 8 minutes after mixing, to a sticky taffy phase until 15 minutes and later a moldable phase where it can be packed to fill bone defects. To assure non-toxicity, the adhesive was chemically analyzed in pre-polymerized and polymerized states to quantify the presence of residual monomers and extracts. The results were all within the acceptable limits.
Biomimetic adhesives
Nature has been developing adhesives for millions of years for the most variable purposes. Protection from predators in animals, self-healing in plants and attachment to surfaces in microorganisms are some examples.Finding inspiration in nature, researchers have been developing and improving adhesives based on natural processes aiming to achieve their undeniable qualities. Over the past decade, the growing knowledge and understanding of biomimetic mechanisms combined with advances in synthesis and polymer design led research onto a new dynamic era where promising results in various groups are finally meeting the needs.
Blue mussels
Blue mussels, shown in Fig. 7, are small bivalves capable of attaching to a wide range of surfaces such as rocks, wood and ship hulls with high strength even at the intertidal line. This is a factor that attracted the attention of researchers as these organisms have developed, in their evolution, solutions to the surface bonding problems underwater.46 | ||
| Fig. 7 Blue mussels attached to a rock on the coast.47 | ||
Blue mussels can, in fact, attach to virtually all types of organic and inorganic surfaces, even the so called adhesion-resistant materials like poly(tetrafluoroethylene) (PTFE).48 Their adhesion mechanism relies on the byssus, an exogenous structure that consists of a stem, multiple byssal threads and byssal plaques, that allows them to fix to surfaces.49,50 Underwater organisms overcome the wet adhesion challenge by the displacement of surface bond ions and ligands through the secretion of particular proteins onto the interface.51 By analyzing their amino acid content, it was found that there is a repetition of the 3,4-dihydroxyphenyl-alanine (DOPA) residue. Further work revealed that this residue is responsible for the mechanical properties of the byssal threads and the formation of the plaque-surface adhesion.52 Its role had also been extensively studied for several years, creating high expectations for its use for medical adhesives.53,54
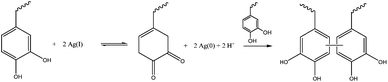
Despite all the information, the true mechanisms of mussel-protein adhesion are not fully understood so many efforts are being made to reveal the outline of such mechanisms. It is believed that DOPA oxidation initiates the reaction. As explained in Fig. 8, the hydroxyl groups are deprotonated under oxidative conditions, turning dopamine into dopaquinone. This molecule suffers rearrangement via intramolecular cyclization to leukodopaminechrome. Further oxidation leads to the formation of 5,6 dihydroxyindole and after this, there are two pathways to form polydopamine. The major reaction is covalent coupling beginning to 2,2′-dihydroxyindole dimer formation and consequently, further polymerization occurs. The minor reaction, the second pathway, is self-assembly between dopamine and the dihydroxyindole.48,55
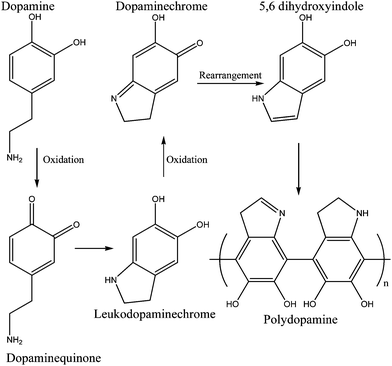 | ||
| Fig. 8 Dopamine crosslinking model—major reaction. Oxidation and further rearrangements lead to the formation of crosslinked dopamine which is structurally similar to melanin.48 | ||
Another model suggests that three key stages are involved in the process. It begins with Fe3+ bringing three catechol chains of proteins that contain DOPA together to form the complex [Fe(dopa)3] (Fig. 9).
![[Fe(dopa)3] complex formation with DOPA oxidation and iron reduction that occur immediately leading to semiquinone formation.](/image/article/2013/BM/c2bm00121g/c2bm00121g-f9.gif) | ||
| Fig. 9 [Fe(dopa)3] complex formation with DOPA oxidation and iron reduction that occur immediately leading to semiquinone formation. | ||
DOPA oxidation and iron reduction occur immediately, leading to semiquinone formation. Protein-based radicals are then generated by reaction of the semiquinones with oxygen. Crosslinking occurs through radical–radical coupling enabling the adhesive with cohesive strength. Eventually, bulk adhesion can occur through radical–surface coupling.52 Thus, the adhesive properties of mussels appear to be attributable to a combination of DOPA, iron and oxygen. Still there are other hypothetical models for the adhesion mechanism of blue mussels to different types of surfaces.56,57 The Fe-catechol chelation has also attracted the attention of Taylor58 and Sever et al.,59 who developed work on these ferric catecholate complexes.
Since their adhesiveness is far superior even to that of synthetic adhesives, with an estimated strength between 320 and 750 kPa60, preliminary studies revealed them as a high-possible concept for in vivo medical applications—such as replacing suturing or stapling. Much has been done in this area and will be discussed in the next sections61–64 (see Table 2 for summary).
PEG-base DOPA
Linear and branched DOPA modified PEG hydrogels containing one to four DOPA endgroups were synthesized as shown in Fig. 10. Gelation time was dependent on polymer architecture, type and concentration of oxidizing agent and presence or absence of N-terminal protecting groups. N-Boc protected PEG-DOPA crosslinked via phenol coupling and quinine methide tanning pathways, whereas oxidation of unprotected PEG-DOPA crosslinked via a pathway that resembled mammalian melanogenesis.69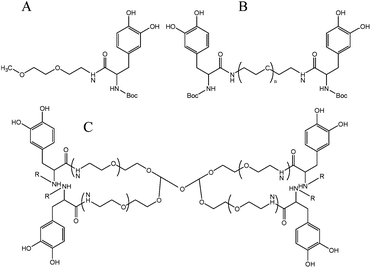 | ||
| Fig. 10 Structure of the different DOPA modified PEG hydrogels. (A) linear mono-functionalized DOPA, (B) linear di-functionalized DOPA and (C) branched. R = Boc or R = H.69 | ||
In 2007, the same PEG-DOPA polymer structure was used in combination with sodium periodate liposomes to create a liquid adhesive precursor that can be stored at room temperature and be thermally activated at the wound site (37 °C). Lap-shear tensile test measurements on porcine dermal tissue surfaces revealed a bond strength of 35.1 kPa ± 12.5 kPa, which was five times higher than fibrin glue.70
The same formulation was later tested for bonding and acute cytotoxicity on direct contact with foetal membranes for 24 hours. The objective was to assess the possibility of using a different sealant for closure of iatrogenic membrane defects. Despite using sodium periodate, which is strongly irritating, to trigger polymerization, CPEG (DOPA modified) adhesive displayed membrane bonding and toxicity comparable to that of a commercially available fibrin adhesive.71 The adhesive was then subjected to a standardized ex vivo evaluation using elastomeric membranes. The closure test of membrane defects of up to 3 mm showed improved sealant efficiency.72
With the objective of avoiding oxidative crosslinking, which is hypothesized to reduce the adhesive properties of DOPA, N-methacrylated DOPA monomers were synthesized and copolymerized with PEG diacrylate using UV or visible light (Fig. 11). Addition of the DOPA monomers to the PEG-DA precursor solution caused a decrease in gel conversion which was dependent on DOPA monomer concentration and photopolymerization initiating system. The extent of DOPA incorporation and elastic modulus also decreased with increasing DOPA concentration; its properties, nevertheless allow its use in many biomedical applications.62
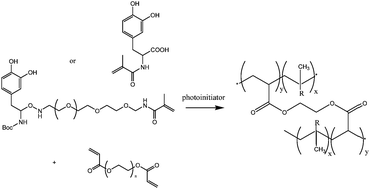 | ||
| Fig. 11 Reaction mechanism between the DOPA modified monomers A or B and the PEG-diacrylate to form a crosslinked network.62 R represents the DOPA modified monomers in the polymer network. | ||
DOPA-modified triblock copolymers with two functional vinyl groups (Fig. 12) were synthesized and used to form adhesive hydrogels by photopolymerization. The block copolymer design facilitated rapid free-radical photopolymerization and high DOPA content compared to previously reported DOPA hydrogels. A contact mechanics method was developed to probe the bulk physical properties and work of adhesion to a titanium surface in aqueous buffer. Work of adhesion to titanium surfaces was reduced by DOPA oxidation and was increased with increasing DOPA content. Moduli of 30–40 kPa was exhibited by the hydrogels which is similar to that of soft tissues.57
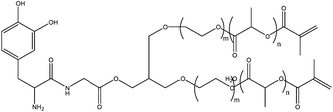 | ||
| Fig. 12 DOPA modified monomer with two vinyl functional groups that allow homopolymerization.57 | ||
With the work developed on DOPA-containing hydrogels, Lee et al. focused on the micro scale and reported a study of the substrate and oxidation dependent adhesive properties of single DOPA molecules.73 For the tips modification, a large amount of methoxy-PEG was used in order to ensure a single DOPA residue was obtained. The contact between a single DOPA residue and a wet metal oxide surface revealed a high strength yet fully reversible, noncovalent interaction. Oxidation of DOPA, as previously reported, dramatically reduced the adhesion strength to metal oxide but when interacting with organic surfaces a strong irreversible covalent bond is formed, view Fig. 13.
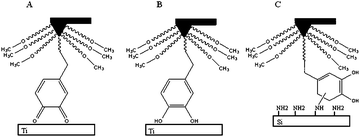 | ||
| Fig. 13 Adhesion profile in (A) titanium surface under oxidizing conditions, (B) titanium surface and (C) organic surface (Silica).73 | ||
Exploiting the mussel's ability to attach to PTFE, the same group developed a two step method of surface modification. Consisting of dipping the material in a dopamine (DN) solution, it allows surface modification of every type of material.48
Aiming at the assessment of tissue adhesives performance and reliability, the group used again a DOPA modified 4-arm PEG adhesive (Fig. 14). In vivo evaluation was performed in a new murine model with induced diabetes. Islets were immobilized onto intra-abdominal surfaces using a tissue adherent hydrogel membrane. A nearly 90% success rate of normoglycemia was reported and minimal inflammatory response indicated the efficacy and safety of this cPEG adhesives. Moreover, one year after transplantation, reliable interfacial maintenance between islet and supporting tissue was observed, which allowed efficient normoglycemic recovery and graft revascularization.64
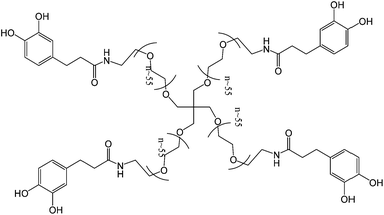 | ||
| Fig. 14 A catechol functionalized PEG hydrogel that was used to test a new paradigm in extrahepatic islet transplantation on a murine model with induced diabetes. No sutures were used and the adhesive proved its efficacy even one year after transplantation.64 | ||
pH responsive self-healing polymeric hydrogels were prepared through dynamically reversible complexation of cPEG with 1,3 benzenediboronic acid. At alkaline pH it demonstrated a covalent gel behavior which dissociated into a viscous liquid at neutral and acidic pH.74
Considering a different polymeric configuration, Lee et al. developed DOPA-tethered PEG adhesives that exhibited better adhesion than end-functionalized PEG-DOPA. PEG-g-catechol was prepared by the step-growth polymerization of PEO to which dopamine was conjugated.75
In order to achieve a major property for biomedical applications, biodegradability, an enzymatically degradable adhesive was described in 2011. The method involves the modification of a branched PEG with DOPA-mimetic catechol linked through an Ala–Ala dipeptide substrate as it possible to observe in Fig. 15. This substrate is degraded by the proteolytic activity of a neutrophil elastase, view Fig. 16. Under oxidizing conditions, gelation and tissue adhesion occurred in less than one minute. Lap shear test revealed adhesion strength of 30.4 ± 3.39 kPa. Adhesive degradation was not shown during short time in vitro analysis; however, in vivo degradation confirmed not only a slow degradation over months but also minimal inflammatory tissue response.66
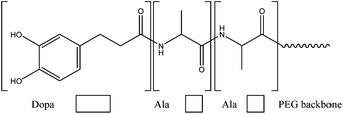 | ||
| Fig. 15 Incorporation of Ala–Ala dipeptide substrate in a catechol modified PEG polymer to allow proteolytic degradation through the action of a neutrophil elastase.66 | ||
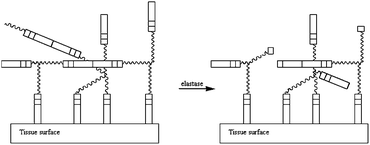 | ||
| Fig. 16 Scheme of the adhesive degradation in the presence of elastase. The enzyme breaks the links between the Ala–Ala sequence in order to degrade the crosslinked network.66 | ||
Recently, this formulation was the base for the development of a silver releasing antibacterial hydrogel. Such a property was achieved through the incorporation of silver that formed nanoparticles and simultaneously served as the oxidizing agent to induce crosslinking, as observed in Fig. 17. The silver release was sustained for at least two weeks and bacterial growth was inhibited without significantly affecting mammalian cell viability.76
 | ||
| Fig. 17 Reaction scheme for hydrogel crosslinking using silver which forms not only the nanoparticles that are going to release silver over time, conferring a antibacterial property to the hydrogel, but also oxidizes the catechol group, allowing for quinine-initiated radical coupling to catechols.76 | ||
Natural backbone
Testing the adhesion strength of mussel protein extracts Ninan et al. had reported a maximum value of 330 kPa after 24 hours of curing, using porcine skin as substrate.65 The same group also studied the strength and curing rate of those extracts on porcine small intestinal submucosa. The joints formed using MAPs were stronger than those formed by butyl and octyl cyanoacrylates (two commercial synthetic adhesives). Still, bonds formed by ethyl cyanoacrylate were even stronger.77As protein separation is very expensive and impractical—about 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mussels are needed to extract 1 g of mussel adhesion proteins (MAPs), alternative strategies are being explored. For instance, co-polypeptides of 3,4-dihydroxyphenyl-alanine and L-lysine were produced to mimic the marine adhesive protein where the ferric ion was the additional crosslinking agent. The higher DOPA content benefits bonding and synchronously, tensile strength is increased with the increase of the molecular weight of the co-polypeptide, likely due to increased intermolecular cohesion. Tensile tests were performed both in dry and wet conditions. The presence of moisture had a significant effect on the adhesive's performance. The highest strength of 208 kPa on dry porcine skin was achieved with 12 hours of curing.56
000 mussels are needed to extract 1 g of mussel adhesion proteins (MAPs), alternative strategies are being explored. For instance, co-polypeptides of 3,4-dihydroxyphenyl-alanine and L-lysine were produced to mimic the marine adhesive protein where the ferric ion was the additional crosslinking agent. The higher DOPA content benefits bonding and synchronously, tensile strength is increased with the increase of the molecular weight of the co-polypeptide, likely due to increased intermolecular cohesion. Tensile tests were performed both in dry and wet conditions. The presence of moisture had a significant effect on the adhesive's performance. The highest strength of 208 kPa on dry porcine skin was achieved with 12 hours of curing.56
A chitosan-based water resistant adhesive following not mussel adhesion but insect sclerotization, using tyrosinase to oxidize dopamine (3,4-dihydroxyphenethylamine).67 The basic mechanism is also quinine adhesion. Shear tests were performed on glass slides after immersion of the joints for 24 hours. An adhesion strength of 450 kPa was achieved in the best-case scenario.
Hyaluronic acid conjugated with dopamine (HA-DN) was mixed with thiol end-capped Pluronic F127 copolymer (Plu-SH) to produce an in situ forming hydrogel based on Michael-type catechol-thiol addition reaction (Fig. 18). The hydrogel was injected as a solution at room temperature but immediately formed a gel at body temperature. A tension test was used to evaluate the adhesion strength using mouse skin as a substrate and an increase of 168% in adhesion force was shown with HA-DN which is coherent with the expected role of the catechol moieties in tissue adhesion.68
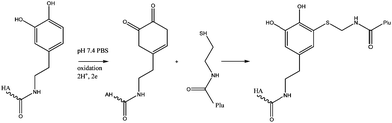 | ||
| Fig. 18 Mechanism of hydrogel formation. Michael-type addition between a quinone form of a catechol group on HA-DN and a thiol group on Plu-SH.68 | ||
Later, a tissue adhesive with catechol functionalized chitosan and thiol-terminated Pluronic F127 (structure in Fig. 19) was developed by Ryu et al.14 Adhesiveness was evaluated using mouse subcutaneous tissue and maximum strength achieved had a value of 15.0 ± 3.5 kPa. Further evaluation of haemostatic properties revealed an excellent haemostatic ability by forming adhesive hydrogel pads on the bleeding site.
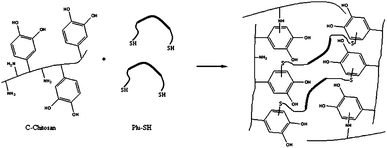 | ||
| Fig. 19 Adhesive formation using catechol functionalized chitosan and thiol functionalized Pluronic F127. A dense polymeric network is developed not only by the crosslinking between C-chitosan and Plu-SH but also between reactive groups in the chitosan chain.14 | ||
Other
DOPA was incorporated into the midblock of poly(methyl methacrylate)—poly(methacrylic acid)—poly(methyl methacrylate) triblock copolymers to form an hydrogel in situ. Its self-assembly ability was obtained by vapor phase solvent exchange from water miscible solvents. As water replaced the solvent, the hydrophobic monomers aggregate and the water-soluble PMAA bridged those aggregates to form an elastic network. The adhesive and mechanical properties were tested using an indentation method and shear moduli between 1 and 5 kPa and interfacial fracture energy of 2 J m−2 were found.78Later, a membrane contact experiment to assess adhesive strength was developed and the previous DOPA modified triblock methacrylic hydrogels were tested along with diblock Boc-DOPA terminated PS-PEO. To measure the adhesive strength, the pressure needed to separate the DOPA functionalized membrane from the substrate (TiO2 and tissue surface) was used as a metric. The results confirm the findings of previous experiments regarding the influence of oxidation.79
In another study where the adhesive strength and fouling ability of DOPA were confirmed, Fan et al. used for the first time a catecholic initiator for surface-initiated polymerization (SIP) to create antifouling polymer coatings on metal surfaces. The approach relies on the use of a bifunctional initiator that contains a catechol end group for surface anchoring and alkyl bromine that allows the activation of SIP (Fig. 20). Atom transfer radical polymerization is then performed to polymerize methyl methacrylate macromonomers with oligo(ethylene glycol) side chains. Cell adhesion was substantially reduced on Ti and 316L stainless steel surfaces where the polymer was grafted compared to unmodified ones.80
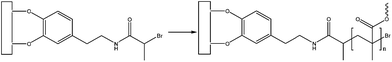 | ||
| Fig. 20 Structure of the bifunctional initiator. DOPA mimetic endgroups allow surface anchoring while the alkyl bromine that activates surface-initiated polymerization.80 | ||
Focusing on increasing biocompatibility of urethane substrates, You et al. conjugated dopamine with a heparin derivative (hepamine), onto a heparin backbone to develop a robust coating. The hepamine-coated poly(urethane) substrate showed significant inhibition of blood coagulation and platelet adhesion. The use of this coating for surface modification shows a number of advantages among which are: no chemical pretreatment of the substrates is necessary, and surface functionalization is a simple, one-step procedure.81
Sandcastle worm
Similar to the blue mussels, other marine organisms depend on the production of adhesive secretions. Another example, the sandcastle worm, shown in Fig. 21, is a reef-forming marine polychaete worm that lives in protective tubular shelters comprised of sand grains and shell fragments, glued together using their own secreted proteins.46,82 | ||
| Fig. 21 Sandcastle worm and its tubular shelters made of sand grains and shell fragments.83 | ||
Such proteins were also separated and analyzed and it was found that, as with blue mussels, DOPA is also an adhesion promoter.84 Furthermore, a high concentration of phosphoserine (∼30 mol%) (structure presented in Fig. 22) indicated its possible participation in the mechanism. Even though they are aquatic organisms, sandcastle worms deliver this underwater glue through a fundamentally different strategy.
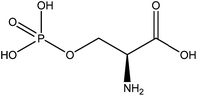 | ||
| Fig. 22 Chemical structure of phosphoserine, a residue present in high concentration in the secreted glue of the sandcastle worm. | ||
It is suggested that the structure of the glue is formed from two interspersed aqueous phases: a dilute equilibrium phase and a denser coacervate phase.85 The sandcastle worm glue is secreted as a colloidal suspension with relatively low interfacial tension and viscosity, allowing it to spread over the substrate surface.86 The glue hardens quickly and becomes a brown and weight bearing solid over several hours. As the secretory system of the sandcastle worm has a pH value of about 5 and the pH of the surrounding seawater is round 8, it is believed that this difference may be a trigger of phase transition.84
Divalent cations, such as Ca2+ and Mg2+, are also believed to take part in the adhesion together with up to 16 charged proteins.85,87 The major contributors to adhesion, 94% of the glue proteins, belong to three different groups: Glycine, Tyrosine-rich; Histidine-repeat and Serine, Tyrosine-rich.86 The presence of lysine and DOPA is once more coherent with their assumed role in underwater adhesion. To assess the bond strength of this natural glue, a pull-out force test was performed and the result achieved was around 300 kPa which also agrees with the values found in literature.60,88
Different approaches to synthesize analogs to the sandcastle worm glue have been explored (Table 3). For instance, a polyacrylate protein glue analog to the secretion of P. californica was developed using two protein analogs that when mixed formed a coacervate.60 The first one comprises dopamine and methacrylamide and the second mimics lysine side chains using N-83-amino-propyl methacrylamide hydrochloride. Mechanical tests performed on bone that was immersed in PBS showed increasing bond strength with increasing divalent cation concentration (Ca2+ and Mg2+ were used) where the maximum was around 100 kPa. Another analog, synthesized through non-gelling collagen and aminated gelatin, was tested in craniofacial reconstruction. The adhesive was non-toxic, degradable, osteoconductive and held its position on normal bone for up to 4 weeks.87
| Composition | Adhesion test | Strength | Ref. |
|---|---|---|---|
| Natural extracts | Pull out force | 300 kPa | 60, 88 |
| Dopamine and methacrylamide + N-83-amino-propyl methacrylamide hydrochloride | Tensile | 100 kPa | 60 |
| 2-(Methacryloyloxy) ethyl phosphate dopamine methacrylamide + poly (acrylamide-co-amino-propyl methacrylamide) + PEG-DA | Tensile | 1.2 MPa | 89 |
More recently, in order to increase the bond strength of complex coacervates that mimic the sandcastle worm secreted glue, Kaur et al. introduced a second polymer network, formed by polymerizable PEG diacrylate (PEG-DA) monomers, within the coacervated copolyelectrolyte network. Crosslinking of the coacervate network occurred through the o-dihydroxyphenyl side chains of 2-(methacryloyloxy) ethyl phosphate dopamine methacrylamide and the amine side chains of poly (acrylamide-co-amino-propyl methacrylamide). Ca2+ was also added to the solution as happened with all similar approaches. After 24 hours of curing, fully submerged in water, mechanical tests revealed a maximum shear bond strength of nearly 1.2 MPa. Higher concentration of PEG-DA increased the bond strength.89
An in vitro test was lately developed to evaluate the ability of this type of adhesives to seal iatrogenic defects. A fetal membrane patch in conjunction with a PEG-DA adhesive and nanosilica-filled coacervates to mimic the sandcastle worm bioadhesive, were used. Toxicity was assessed through direct contact with human fetal membranes in an organ culture setting. Non-toxicity and sealing ability seem to make this a possible strategy to prevent iatrogenic preterm premature rupture of fetal membranes.90
Barnacles
Barnacles (Fig. 23) are marine crustaceans that, when adults, attach to different types of substrates depending on their subspecies.91 In order to attach themselves, they use a mechanism that is similar to the complex coacervation that is also found in the sandcastle worm. | ||
| Fig. 23 Acorn barnacles attached to a rock at low tide.91 | ||
It is believed that these organisms secrete a proteinaceous adhesive just a few microns thick. The details of how the proteins are mixed to form the glue, or the trigger that initiates solidification, are still unknown. However, several groups have been analyzing the properties and possible adhesion mechanisms. First, some authors differentiate the adhesion of adult barnacles and cyprids (a larval stage before maturation) as well as temporary and permanent adhesion.92 Second, there is no agreement as to whether secondary covalent curing does or does not happen.46,92 Focusing on adult barnacles, the cement is produced in giant cells and then transported through ducts to the periphery of the baseplate.46 Initially it is clear and has low viscosity but solidifies and becomes insoluble. After it is fully cured it is extremely hard to solubilize it and it withstands ruthless denaturing conditions. From some of these experiments, it is implied that there is disulfide bond formation in cohesion.93
Regarding its composition, two major polypeptides (100 kDa and 52 kDa), highly insoluble and hydrophobic are considered to form structural proteins, providing a spontaneously self-assembling framework upon release in favorable conditions. Other peptides, less abundant, have been also found, one acting as a surface primer (68 kDa) and another supposedly involved in calcite binding. Unlike Blue mussels, there is no evidence for metal binding94 or DOPA content.50
Considering temporary cyprid adhesion, not only is the proteinaceous glue presumed to act as a “wet” adhesive as the cuticular villi may contribute to adhesion, as with the spatulae of geckos (fibrillar adhesion).92 For permanent adhesion, a debate on the relation of cyprid and adult cement has now ceased as Aldred et al. showed morphological differences between them.95 The hypothesis of cement curing via quinine crosslinking is supported by the identification of diphenols and the enzyme polyphenoloxidase in the cement apparatus.96
A study with a synthetic peptide derived from a repeating unit of the 20 kDa barnacle cement protein suggests a pH jump as a trigger to initiate solidification. The peptide that was soluble at low ionic strength self-assembled irreversibly into a mesh of insoluble fibrils.97 In spite of knowing of the high adhesion strength to different substrates, there are still many questions to be answered before it can be used in the biomedical field.
Spider net
Moving from the underwater organisms, others have attracted scientists’ attention. Here the focus lays on the orb-weaving spiders and their highly evolved adhesive mechanism to capture prey. The unique properties of the spider silk and the many possibilities of its use for biomedical applications98 are as fascinating as the glue laid on the silk threads is considered one of the most effective biological ones.99Considering its chemical composition, high concentrations of water-soluble organic compounds related to neurotransmitters, small peptides, free amino acids and low concentrations of inorganic salts and glycoproteins were found.100 However, in-depth knowledge of its molecular structure is lacking.
Recent studies reveal the adhesiveness as a result of an extension rate-dependent behavior of the glue droplets that are composed of an aqueous coat of salt surrounding microscopic nodules made of glycoproteins.101 When the silk faces high extension rates, as in the capturing of prey, the glue becomes highly viscous, thus providing maximum adhesion. At slow extension rates, like the prey's movements to escape the web, the glue droplets deform like an elastic rubber band-like material to keep it trapped. Although a key role is attributed to the glycoproteins, the molecular components that cause this change in viscoelasticity remain unknown.102
The information on the spider net adhesion mechanism is still quite scarce but the work that has been undertaken is only the start and future investigations will provide more information that will allow its use in biomaterials design.
Gecko
Geckos are other organisms highly dependent on a completely different adhesion mechanism based on physical properties. These lizards’ adhesive strategy relies on their foot pads (Fig. 24) composed of keratinous foot-hairs called setae that then are subdivided into spatula of around 200 nm.13 The physical arrangement allows attachment onto smooth vertical or even inverted surfaces. Although strong, the adhesion is temporary, permitting detachment and reattachment during locomotion. The first direct measurements of single setae force were reported by Autumn et al. where a 2D micro electro mechanical system (MEMS) was used. The study revealed that a seta was ten times more effective at adhesion than predictions from maximal estimates on whole animals and the adhesive force values supported the hypothesis that individual seta operate by van der Waals forces.103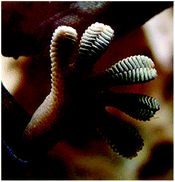 | ||
| Fig. 24 Gecko's foot as it walks on a glass surface. These lizards rely on physical adhesion to cling onto vertical and even inverted surfaces. | ||
These properties caught the attention of many researchers trying to develop synthetic approaches to mimic the nanoscale setae surface features. In 2003, Geim et al. opened a door onto this area with the gecko tape. It consisted of the microfabrication of dense arrays of flexible polyimide pillars with an optimized geometry to ensure their collective adhesion.104 Although its force was expected to stick a person to a ceiling by only one hand, the adhesive's properties were compromised after a few cycles.
Others worked on similar approaches105–107 but the same problems were found: loss of adhesive properties over many contact cycles108 and in wet conditions.109 In order to maintain performance over many cycles and specially upon immersion in water, Lee et al. developed a hybrid biologically inspired adhesive combining geckos and mussels. The adhesive consisted of an array of nanofabricated pillars coated with a thin polymer layer that mimicked the adhesive proteins found in blue mussels.13 Performance was maintained for over a thousand contact cycles in both dry and wet conditions. A nearly 15-fold increase in wet adhesion was observed with the coated pillars.
In 2008 a biodegradable and biocompatible tissue adhesive was reported. A poly(glycerol-co-sebacate acrylate) was used and its surface modified to mimic the gecko's feet nanotopography.15 The nanoscale pillar dimensions were varied to optimize tissue adhesion. Dextran coating increased interfacial adhesion strength both in vitro (porcine intestine) and in vivo (rat abdominal subfascial environment). Such results looked very promising regarding its application in wound closure.
Recently, Zhang et al. grafted dopamine onto a polyacrylate backbone to mimic a gecko nano-structural adhesive.110 The incorporation of dopamine resulted in better wettability and hydrophobicity and tensile strength increased from 53 to 124 kPa in its presence. Nano-patterns were formed on the adhesion interface during bonding with a diameter of around 100 nm in each contact point. No toxicity experiments were done to evaluate its possible use in wound healing.
Despite its interesting features, there are still improvements to be done in this field. Fabrication of these patterned adhesives is still expensive and increases in strength are also needed.
Summary
Various strategies have been sought in the last decade to face the current demands in wound closure. The main advantages and disadvantages of tissue adhesives, both commercially available and those in research are presented in Table 4, as well as degradation rates and current applications.| Type | Degradation | Advantages | Disadvantages | Application | |
|---|---|---|---|---|---|
| Biomimetic | Mussel | Variable depending on formulation backbone | Biocompatible | Synthetic route | In development |
| Sandcastle worm | Biodegradable | High cost | |||
| Barnacles | Independent from surface preparation | Lower strength | |||
| Spider net | |||||
| Gecko | |||||
| Synthetic/semi-synthetic | Cyanoacrylates | ∼8 weeks | Controlled properties | Cytotoxic | Skin, cornea, gastric therapy, laparoscopic surgery, dura mater, cardiovascular surgery, cartilage, bone |
| PEG-based | 4–12 weeks | Easy preparation | |||
| Urethane-based | Non-degradable | High strength | |||
| Low cost |
Future perspectives
With the rapid development in biomedical technology and ever growing knowledge in biological mechanisms, tissue adhesives may offer now unthinkable possibilities. Not only closure but also healing promotion and tissue reconstruction are included. Besides the current models of marine mussels, sandcastle worms and others, there is much still to be learned about nature. An animal’s and plant's ability to adhere to surfaces in moist environments and resist the strong adversities that can easily cause detachment of synthetic adhesives continues to intrigue researchers throughout the world.Functionalization is now the key word in wound adhesives. Strategies using the attachment of functional adhesive groups to polymer backbones have been providing promising results and are leading research as better conditions are being created.
Acknowledgements
The authors wish to thank Professor Abhay Pandit for his valuable comments and ideas to the writing of this review and the ERASMUS Programme for financial support during the exchange period.Notes and references
- J. H. Breasted, The Edwin Smith Surgical Papyrus: Hieroglyphic Transliteration, Translation and Commentary, Kessinger Publishing, 2006 Search PubMed.
- A. Lauto, M. Damia, L. Foster and R. John, J. Chem. Technol. Biotechnol., 2008, 83, 464–472 CrossRef CAS.
- K. Hida, S. Yamaguchi, T. Seki, S. Yano, M. Akino, S. Terasaka, T. Uchida and Y. Iwasaki, Surg. Neurol., 2006, 65, 136–142 CrossRef.
- K. D. H. Murray, C.-H. Hsia, J.-Y. James and A. G. Little, Chest, 2002, 122, 2146–2149 CrossRef.
- R. L. Reed, R. C. Merrell, W. C. Meyers and R. P. Fischer, Ann. Surg., 1992, 216, 524–538 CrossRef.
- S. Siemer, S. Lahme, S. Altziebler, S. Machtens, W. Strohmaier, H.-W. Wechsel, P. Goebell, N. Schmeller, R. Oberneder, J.-U. Stolzenburg, H. Becker, W. Lüftenegger, V. Tetens and H. Van Poppel, Eur. Urol., 2007, 52, 1156–1163 CrossRef CAS.
- A. Mattick, G. Clegg, T. Beattie and T. Ahmad, Emerg. Med. J., 2002, 19, 405–407 CrossRef CAS.
- J. Quinn, A. Drzewiecki, M. Li, I. Stiell, T. Sutcliffe, T. Elmslie and W. Wood, Ann. Emerg. Med., 1993, 22, 1130–1135 CrossRef CAS.
- M. G. Orlinsky, R. M. Goldberg, L. Chan, A. Puertos and H. L. Slajer, Am. J. Emerg. Med., 1995, 13(1), 77 CrossRef CAS.
- M. H. Osmond, T. P. Klassen and J. V. Quinn, J. Pediatr., 1995, 126, 892–895 CrossRef CAS.
- a. m. t. MedMarket Diligence.
- I. Global Industry Analysts.
- H. Lee, B. P. Lee and P. B. Messersmith, Nature, 2007, 448, 338–341 CrossRef CAS.
- J. H. Ryu, Y. Lee, W. H. Kong, T. G. Kim, T. G. Park and H. Lee, Biomacromolecules, 2011, 12, 2653–2659 CrossRef CAS.
- A. Mahdavi, L. Ferreira, C. Sundback, J. W. Nichol, E. P. Chan, D. J. D. Carter, C. J. Bettinger, S. Patanavanich, L. Chignozha and E. Ben-Joseph, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2307 CrossRef CAS.
- M. T. Ryou and C. Christopher, Techniques in Gastrointestinal Endoscopy, 2006, 33–37 CrossRef.
- Y.-C. Tseng, Y. Tabata, S.-H. Hyon and Y. Ikada, J. Biomed. Mater. Res., 1990, 24, 1355–1367 CrossRef CAS.
- C. P. K. Sundaram and C. Alison, Indian Journal of Urology, 2010, 26, 374–378 CrossRef.
- P. A. S. Leggat, R. Derek and U. Kedjarune, ANZ J. Surg., 2007, 77, 209–213 CrossRef.
- D. F. Torchiana, Journal of Cardiac Surgery, 2003, 18, 504–506 CrossRef.
- N. Artzi, S. Tarek, C. Crespo, A. B. Ramos, H. K. Chenault and E. R. Edelman, Macromol. Biosci., 2009, 9, 754–765 CrossRef CAS.
- I. Strehin, N. Zayna, K. Arora, T. Nguyen and J. Elisseeff, Biomaterials, 2010, 31, 2788–2797 CrossRef CAS.
- H. Li, N. Rui, J. Yang, J. Nie and D. Yang, Carbohydr. Polym., 2011, 86, 1578–1585 CrossRef CAS.
- P. Ferreira, R. Pereira, J. F. J. Coelho, A. F. M. Silva and M. H. Gil, Int. J. Biol. Macromol., 2007, 40, 144–152 CrossRef CAS.
- P. W. M. Fedak and A. Kasatkin, Surgical Innovation, 2011, 18, NP8–NP11 CrossRef.
- S. Kull, I. Martinelli, E. Briganti, P. Losi, D. Spiller, S. Tonlorenzi and G. Soldani, J. Surg. Res., 2009, 157, E15–E21 CrossRef CAS.
- J. V. Quinn, Tissue Adhesives in Clinical Medicine, BC Decker, 2005 Search PubMed.
- C. Esposito, R. Damiano, A. Settimi, M. De Marco, P. Maglio and A. Centonze, Surg. Endosc., 2004, 18, 290–292 CrossRef CAS.
- U. Narang, L. Mainwaring, G. Spath and J. Barefoot, Journal of Cutaneous Medicine and Surgery: Incorporating Medical and Surgical Dermatology, 2003, 7, 13–19 Search PubMed.
- B. Mizrahi, C. F. Stefanescu, C. Yang, M. W. Lawlor, D. Ko, R. Langer and D. S. Kohane, Acta Biomater., 2011, 7, 3150–3157 CrossRef CAS.
- A. J. Singer and H. C. Thode, Am. J. Surg., 2004, 187, 238–248 CrossRef CAS.
- S. Mizrahi, A. Bickel and E. Ben-Layish, J. Pediatr. Surg., 1988, 23, 312–313 CrossRef CAS.
- F. Sabol, T. Vasilenko, M. Novotný, Z. Tomori, N. Bobrov, J. Živčák, R. Hudák and P. Gál, Eur. Surg. Res., 2010, 45, 321–326 CrossRef CAS.
- S. Seewald, S. Groth, P. V. J. Sriram, H. Xikun, T. Akaraviputh, G. Mendoza, B. Brand, U. Seitz, F. Thonke and N. Soehendra, Gastrointest. Endosc., 2002, 56, 916–919 CrossRef.
- S. Chibbaro and L. Tacconi, J. Clin. Neurosci., 2009, 16, 535–539 CrossRef.
- T. B. W. Bruns and J. Mack, Am. Fam. Phys., 2000, 61, 1383–1388 CAS.
- A. K. Gosain and V. B. Lyon, Plast. Reconstr. Surg., 2002, 110(6), 1581–1584 CrossRef.
- T. M. Shazly, N. Artzi, F. Boehning and E. R. Edelman, Biomaterials, 2008, 29, 4584–4591 CrossRef CAS.
- R. Fasol, T. Wild and S. El Dsoki, Ann. Thorac. Surg., 2004, 77, 1070–1072 CrossRef.
- A. N. Azadani, B. M. Peter, L. Ge, Y. Shen, C.-S. Jhun, T. S. Guy and E. E. Tseng, Ann. Thorac. Surg., 2009, 87, 1154–1160 CrossRef.
- M. A. Serban, B. Panilaitis and D. L. Kaplan, J. Biomed. Mater. Res., Part A, 2011, 98A, 567–575 CrossRef CAS.
- T. Lipatova, Adv. Polym. Sci., 1986, 65–93 CrossRef CAS.
- A. Duarte, J. Coelho, J. Bordado, M. Cidade and M. Gil, Prog. Polym. Sci., 2011, 00, 000 Search PubMed.
- P. Ferreira, A. Silva, M. Pinto and M. Gil, J. Mater. Sci.: Mater. Med., 2008, 19, 111–120 CrossRef CAS.
- P. Ferreira, J. Coelho and M. Gil, Int. J. Pharm., 2008, 352, 172–181 CrossRef CAS.
- R. J. Stewart, T. C. Ransom and V. Hlady, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 757–771 CrossRef CAS.
- A. w. p.-n. d. Trepte, 2007.
- H. D. Lee, M. Shara, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS.
- H. R. Silverman and F. Franscisco, Mar. Biotechnol., 2007, 9 Search PubMed.
- Biological Adhesive Systems, From Nature to Technical and Medical Application, ed. I. G. Janek von Byern, Springer, Wien, New York, Austria, 1st edn, 2010 Search PubMed.
- J. W. Costerton, The Biofilm Primer, Springer, Berlin, 2007 Search PubMed.
- J. J. Wilker, Angew. Chem., Int. Ed., 2010, 49, 8076–8078 CrossRef CAS.
- J. H. Waite and M. L. Tanzer, Science, 1981, 212, 1038–1040 CAS.
- M. Yu, J. Hwang and T. J. Deming, J. Am. Chem. Soc., 1999, 121, 5825–5826 CrossRef CAS.
- S. Hong, Y. S. Na, S. Choi, I. T. Song, W. Y. Kim and H. Lee, Adv. Funct. Mater., 2012, 22, 4711–4717 CrossRef CAS.
- J. Wang, C. S. Liu, X. Lu and M. Yin, Biomaterials, 2007, 28, 3456–3468 CrossRef CAS.
- B. P. Lee, C. Y. Chao, F. N. Nunalee, E. Motan, K. R. Shull and P. B. Messersmith, Macromolecules, 2006, 39, 1740–1748 CrossRef CAS.
- S. W. Taylor, D. B. Chase, M. H. Emptage, M. J. Nelson and J. H. Waite, Inorg. Chem., 1996, 35, 7572–7577 CrossRef CAS.
- M. J. Sever, J. T. Weisser, J. Monahan, S. Srinivasan and J. J. Wilker, Angew. Chem., 2004, 116, 454–456 CrossRef.
- H. Shao, K. N. Bachus and R. J. Stewart, Macromol. Biosci., 2009, 9, 464–471 CrossRef CAS.
- H. J. Cha, D. S. Hwang and S. Lim, Biotechnol. J., 2008, 3, 631–638 CrossRef CAS.
- B. P. Lee, K. Huang, F. N. Nunalee, K. R. Shull and P. B. Messersmith, J. Biomater. Sci., Polym. Ed., 2004, 15, 449–464 CrossRef CAS.
- K. Huang, B. P. Lee, D. R. Ingram and P. B. Messersmith, Biomacromolecules, 2002, 3, 397–406 CrossRef CAS.
- C. E. Brubaker, K. Hermann, L.-J. Wang, D. B. Kaufman and P. B. Messersmith, Biomaterials, 2010, 31, 420–427 CrossRef CAS.
- L. Ninan, J. Monahan, R. L. Stroshine, J. J. Wilker and R. Shi, Biomaterials, 2003, 24, 4091–4099 CrossRef CAS.
- C. E. Brubaker and P. B. Messersmith, Biomacromolecules, 2011, 12, 4326–4334 CrossRef CAS.
- K. Yamada, T. Chen, G. Kumar, O. Vesnovsky, L. D. T. Topoleski and G. F. Payne, Biomacromolecules, 2000, 1, 252–258 CrossRef CAS.
- Y. Lee, H. J. Chung, S. Yeo, C.-H. Ahn, H. Lee, P. B. Messersmith and T. G. Park, Soft Matter, 2010, 6, 977–983 RSC.
- B. P. Lee, J. L. Dalsin and P. B. Messersmith, Biomacromolecules, 2002, 3, 1038–1047 CrossRef CAS.
- S. A. Burke, M. Ritter-Jones, B. P. Lee and P. B. Messersmith, Biomed. Mater., 2007, 2, 203–210 CrossRef CAS.
- G. Bilic, C. Brubaker, P. B. Messersmith, A. S. Mallik, T. M. Quinn, C. Haller, E. Done, L. Gucciardo, S. M. Zeisberger, R. Zimmermann, J. Deprest and A. H. Zisch, Am. J. Obstet. Gynecol., 2010, 202 Search PubMed.
- C. M. Haller, W. Buerzle, C. E. Brubaker, P. B. Messersmith, E. Mazza, N. Ochsenbein-Koelble, R. Zimmermann and M. Ehrbar, Prenatal Diagn., 2011, 31, 654–660 CrossRef CAS.
- H. S. Lee, F. Norbert and P. B. Messersmith, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12999–13003 CrossRef CAS.
- L. He, D. E. Fullenkamp, J. G. Rivera and P. B. Messersmith, Chem. Commun., 2011, 47, 7497–7499 RSC.
- H. Lee, K. D. Lee, K. B. Pyo, S. Y. Park and H. Lee, Langmuir, 2010, 26, 3790–3793 CrossRef CAS.
- D. E. Fullenkamp, J. G. Rivera, Y. Gong, A. K. H. Lau, L. He, R. Varshney and P. B. Messersmith, Biomaterials, 2012, 33, 3783–3791 CrossRef CAS.
- L. Ninan, R. L. Stroshine, J. J. Wilker and R. Shi, Acta Biomater., 2007, 3, 687–694 CrossRef CAS.
- M. Guvendiren, P. B. Messersmith and K. R. Shull, Biomacromolecules, 2007, 9, 122–128 CrossRef.
- M. Guvendiren, D. A. Brass, P. B. Messersmith and K. R. Shull, J. Adhes., 2009, 85, 631–645 CrossRef CAS.
- X. Fan, L. Lin, J. L. Dalsin and P. B. Messersmith, J. Am. Chem. Soc., 2005, 127, 15843–15847 CrossRef CAS.
- I. You, S. M. Kang, Y. Byun and H. Lee, Bioconjugate Chem., 2011, 22, 1264–1269 CrossRef CAS.
- R. J. Stewart, Appl. Microbiol. Biotechnol., 2011, 89, 27–33 CrossRef CAS.
- J. L. Garfield, 2010.
- R. J. W. Stewart, S. Ching and H. Shao, Adv. Colloid Interface Sci., 2010, 167, 85–93 CrossRef.
- R. J. Stewart, J. C. Weaver, D. E. Morse and J. H. Waite, J. Exp. Biol., 2004, 207, 4727–4734 CrossRef CAS.
- B. J. Endrizzi and R. J. Stewart, J. Adhes., 2009, 85, 546–559 CrossRef CAS.
- B. D. Winslow, H. Shao, R. J. Stewart and P. A. Tresco, Biomaterials, 2010, 31, 9373–9381 CrossRef CAS.
- C. F. Sun, G. E. Fantner, J. Adams, P. K. Hansma and J. H. Waite, J. Exp. Biol., 2007, 210(8), 1481–1488 CrossRef CAS.
- S. Kaur, G. M. Weerasekare and R. J. Stewart, ACS Appl. Mater. Interfaces, 2011, 3, 941–944 CAS.
- L. K. Mann, R. Papanna, K. J. Moise Jr, R. H. Byrd, E. J. Popek, S. Kaur, S. C. G. Tseng and R. J. Stewart, Acta Biomater., 2012, 8, 2160–2165 CrossRef CAS.
- S. Ballard, www.daviddarling.info/encyclopedia/B/barnacle.html.
- N. Aldred and A. S. Clare, Biofouling, 2008, 24, 351–363 CrossRef CAS.
- K. Kamino, K. Inoue, T. Maruyama, N. Takamatsu, S. Harayama and Y. Shizuri, J. Biol. Chem., 2000, 275, 27360–27365 CAS.
- M. Wiegemann, Aquat. Sci. – Res. Across Boundaries, 2005, 67, 166–176 CAS.
- N. Aldred, I. Y. Phang, S. L. Conlan, A. S. Clare and G. J. Vancso, Biofouling, 2008, 24, 97–107 CrossRef CAS.
- G. Walker, Mar. Biol., 1971, 9, 205–212 CrossRef.
- M. Nakano, J.-R. Shen and K. Kamino, Biomacromolecules, 2007, 8, 1830–1835 CrossRef CAS.
- R. V. Lewis, Chem. Rev., 2006, 106, 3762–3774 CrossRef CAS.
- F. Vollrath, W. J. Fairbrother, R. J. P. Williams, E. K. Tillinghast, D. T. Bernstein, K. S. Gallagher and M. A. Townley, Nature, 1990, 345, 526–528 CrossRef CAS.
- O. Choresh, B. Bayarmagnai and R. V. Lewis, Biomacromolecules, 2009, 10, 2852–2856 CrossRef CAS.
- V. Sahni, T. A. Blackledge and A. Dhinojwala, Nat. Commun., 2010, 1, 19 CrossRef.
- H. Lee, Nature, 2010, 465, 298–299 CrossRef CAS.
- K. Autumn, Y. A. Liang, S. T. Hsieh, W. Zesch, W. P. Chan, T. W. Kenny, R. Fearing and R. J. Full, Nature, 2000, 405, 681–685 CrossRef CAS.
- A. K. Geim, S. V. Dubonos, I. V. Grigorieva, K. S. Novoselov, A. A. Zhukov and S. Y. Shapoval, Nat. Mater., 2003, 2, 461–463 CrossRef CAS.
- M. T. Northen and K. L. Turner, Nanotechnology, 2005, 16, 1159 CrossRef CAS.
- M. Sitti and R. S. Fearing, J. Adhes. Sci. Technol., 2003, 17, 1055–1073 CrossRef CAS.
- B. Yurdumakan, N. R. Raravikar, P. M. Ajayan and A. Dhinojwala, Chem. Commun., 2005, 3799–3801 RSC.
- M. T. Northen and K. L. Turner, Sens. Actuators, A, 2006, 130, 583–587 CrossRef.
- W. Sun, P. Neuzil, T. S. Kustandi, S. Oh and V. D. Samper, Biophys. J., 2005, 89, L14–L17 CrossRef CAS.
- F. Zhang, S. Liu, Y. Zhang, J. Xu and Z. Chi, Int. J. Adhes. Adhes., 2011, 32, 583–586 CrossRef.
| This journal is © The Royal Society of Chemistry 2013 |