Effect of shell-crosslinking of micelles on endocytosis and exocytosis: acceleration of exocytosis by crosslinking†
Yoseop
Kim
ab,
Mohammad H.
Pourgholami
b,
David L.
Morris
b,
Hongxu
Lu
a and
Martina H.
Stenzel
*a
aCentre for Advanced Macromolecular Design (CAMD), University of New South Wales, Sydney, NSW 2052, Australia. E-mail: M.Stenzel@unsw.edu.au; Fax: +61-2-93856250; Tel: +61-2-93854344
bCancer Research Laboratories, Department of Surgery, St. George Hospital, University of New South Wales, Sydney, NSW 2052, Australia
First published on 21st December 2012
Abstract
Uptake of drug-loaded micelles by tumour cell lines can be the crucial step in the creation of an efficient drug delivery system. Crosslinking of micelles has increasingly been proposed as a pathway to create stable nanoparticles. So far, little is known how crosslinking can affect the interaction of these nanocarriers with cells. The aim of this study is therefore to investigate the effect of crosslinking on exo- and endocytosis. RAFT (reversible addition fragmentation chain transfer) polymerization has been used to synthesize the block copolymer poly(methyl methacrylate)-block-poly(polyethylene glycol methyl ether methacrylate) PMMA-b-P(PEGMEMA), which was subsequently self-assembled into micelles of 20 nm. For comparison, the micelles were shell-crosslinked by incorporating methacrylic acid into the shell, which was used as a reactive group for crosslinking with 1,8-diaminooctane. The hydrodynamic diameter of the shell-crosslinked micelle was with 25 nm similar to that of the non-crosslinked one. Endocytosis of both micelles was significantly reduced by the presence of NaN3 or at 4 °C suggesting an energy dependent process. The internalization pathways of the block copolymer micelles in OVCAR-3 cells were elucidated using endocytosis inhibitors. Both nanoparticles, micelles and shell-crosslinked micelles, were internalized by caveolae mediated endocytosis while clathrin mediated endocytosis did not play a noticeable role. Shell-crosslinking therefore did not have an effect on endocytosis. However, a considerable difference was found in the exocytosis of both particles. While the micelle was lodged inside the cell for an extended period of time with less than 3% released in two hours, the shell-crosslinked micelle quickly exited the OVCAR-3 cells (25% in two hours). For comparison, a small molecule (Lucifer yellow) was found to be only marginally faster than the crosslinked micelles (40% in 2 h). These results could have implications on the use polymer–drug conjugates or drug carriers where the drug needs to be released before the polymer undergoes exocytosis.
Introduction
One third of drugs in development are hydrophobic molecules and a significant number of drugs fail in trials because of poor pharmacokinetics.1 Reasons are manifold and range from poor solubility and degradation to fast clearance and drug-resistance. Nano-sized drug delivery systems have been proposed as a solution to many of these issues, especially for the treatment of cancer. The enhanced permeation retention (EPR) effect has often been cited as a possible pathway to passively target nanoparticles to tumours2 although there are some constraints to this idea.3 Another possible advantage of nanoparticles for drug delivery is their potential to overcome multi-drug resistance (MDR). MDR to chemotherapeutic drugs remains a major stepping stone and is often responsible for the failure of treatment. MDR is often associated with over-expression of drug transporters such as P-glycoprotein. Nanoparticles typically enter cells via endocytosis, by-passing the efflux pumps. Nanoparticles are therefore particularly successful when dealing with MDR cell lines.4 Polymeric micelles have been widely utilized as drug delivery systems to overcome these problems. Block copolymer micelles are composed of amphiphilic polymers in which the hydrophobic block forms the core of the micelle and the hydrophilic block builds up the corona resulting in increased overall solubility of hydrophobic drugs, which ultimately affects pharmacokinetics.5–10 Owing to the importance of the pathway of the cellular entry of the drug carrier, a range of studies have been devoted to understanding the endocytotic pathway of micelles, in particular that of PEO–PPO–PEO (poly(ethylene oxide)-b-poly(propylene oxide)-b-poly(ethylene oxide)), an amphiphilic triblock copolymer, also known as Pluronic or poloxamers. The focus of these studies was often the relationship between the stability of the micelle or the critical micelle concentration (CMC) and the amount of cellular uptake.11–14 While the bulk of research has had a strong emphasis on Pluronic, a handful of studies have investigated the interaction of various other micelles with cells. Most commonly, micelles with high Tg cores were employed. Akimoto et al. employed a thermo-responsive micelle with a PLA core to investigate the endocytosis of micelles,15 while Zhao and Yung looked at the uptake efficiency of folate containing micelles.16 A good selection of other examples are available, but in most cases not much attention was paid to the type of uptake and only an overall uptake efficiency was given.17–20 The reader is referred to a review by Maysinger and co-workers, which summarized aspects of the interactions of micelles with cells,1 while Verma and Stellacci reviewed aspects of the interactions of cells with nanoparticles in general.21Micelles are typically taken up by cells via endocytosis. Endocytosis can be divided in to two broad categories: phagocytosis (uptake of large particles, >500 nm) and pinocytosis (uptake of fluids and solutes).22 Based on the type of membrane structure involved in the internalization, pinocytosis can also be divided into clathrin mediated endocytosis and caveolae mediated endocytosis.22,23 Caveolae are flask shaped plasma membrane invaginations with diameters of around 70 nm that function as trafficking vesicles and organized signal transduction compartments. They are formed from hetero oligomers of the structural coat proteins caveolin-1 (CAV1) and caveolin-2 (CAV2).24,25 Caveolae are expressed in most normal organs and many solid tumours, mostly by adipocytes, cardiomyocytes and continuous endothelial and epithelial cells.26 Clathrin mediated endocytosis involves the polymerization of a cytosolic protein, clathrin, which binds to the cell plasma membrane and induces the formation of coated pits with sizes of about 100 nm.26 Numerous studies have focused on the mechanisms and pathways involved in the endocytosis of various types of nanoparticles. No pathway has been singled out and the uptake ranges most commonly from caveolae or clathrin mediated endocytosis to macropinocytosis.27,28 A strong focus in the current literature has been given to the uptake of nanoparticles alone, but not the retention. Inspired by the results of clonogenic assays we estimated that the retention of micelles inside a cell may be an important factor when evaluating drug carriers.29 The surviving cell fractions after 72 hours incubation with an anti-cancer drug, here cisplatin, were isolated resulting in quick re-growth and colony formation. Treated with a cisplatin loaded micelle, the surviving fraction continued to die even when immersed in fresh media.29 It seems therefore, that a drug carrier can lead to a long-lasting result when retained inside the cell.
The objective of the present study was to compare the endocytosis and exocytosis of micelles and shell-crosslinked micelles. Micelles are now established drug delivery vehicles and some selected examples have made it to clinical trials.30 Shell-crosslinked micelles have widely been proposed as pathways to stabilize micelles.31,32 The dynamic structure of micelles can lead to disassembly at low concentrations or when in contact with biological molecules resulting in a low cellular uptake.33,34 The central question is if crosslinking affects either endo- or exocytosis.
A block copolymer, PMMA-b-PPEGMEMA, was prepared using RAFT polymerization. In addition, a similar block copolymer with the same number of repeating units was synthesized with a small percentage of carboxylate units in the water-soluble block for further crosslinking (Scheme 1). The exo- and endocytosis of both micelles, crosslinked (CL) and non-crosslinked, were subsequently studied in vitro using human ovarian cancer cell lines (OVCAR-3 cells).
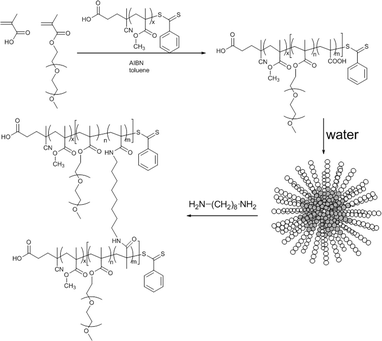 | ||
| Scheme 1 PMMA-b-P(PEGMEMA-co-MAA) block copolymer synthesis via RAFT polymerization using PMMA macroRAFT agent, followed by self-assembly of the block copolymer and shell-crosslinking using 1,8-diaminooctane. The uncrosslinked micelles were obtained using PEGMEMA only (no MAA). | ||
Experimental
Materials
Toluene (Ajax, 99.4%), N,N-dimethyl acetamide (DMAc; Aldrich, 99.9%), dimethyl sulfoxide (DMSO; Ajax, 98.9%), N,N-dimethyl formaldehyde (DMF; Aldrich), diethyl ether (Univar), petroleum spirit (Aldrich), Albendazole (ABZ; Aldrich), sodium azide (Aldrich), sucrose (Ajax), 2-hydroxypropyl-β-cyclodextrin (Aldrich), Lucifer yellow (Aldrich), Filipin(III) (Aldrich) and Nile Red (Aldrich) were used without any further purification. Poly(ethylene glycol) methyl ether methacrylate (PEGMEMA, Mn = 300 g mol−1) (Aldrich, reagent), methyl methacrylate (MMA) (Aldrich, reagent) were destabilized by passing over a column of basic alumina and stored at −7 °C. 2,2-Azobisisobutyronitrile (Fluka, 98%) was purified by recrystallization from methanol. The RAFT agent 4-cyanopentanoic acid dithiobenzoate (CPADB) was prepared according to the procedure described elsewhere.35Synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, δ = 5.7–6.0 ppm) and the –O–CH3 peak (δ = 3.8–4.2 ppm). A conversion of 50% was obtained which is equivalent to 50 MMA repeating units.
CH2, δ = 5.7–6.0 ppm), P(PEGMEMA) –CH2–CH2 peak (δ = 3.8–4.2 ppm) and block copolymer PMMA–b-P(PEGMEMA)–CH2–CH2 peak (δ = 0.8–1.2 ppm).
CH2, δ = 5.7–6.0 ppm), P(PEGMEMA) –CH2–CH2 peak (δ = 3.8–4.2 ppm) and block copolymer –CH2–CH2 peak (δ = 0.8–1.2 ppm).
CH2, δ = 5.7–6.0 ppm) and the –O–CH3 peak (δ = 3.8–4.2 ppm). A conversion of 50% was obtained which is equivalent to 50 MMA repeating units.
CH2, δ = 5.7–6.0 ppm), P(PEGMEMA) –CH2–CH2 peak (δ = 3.8–4.2 ppm) and block copolymer PMMA–b-P(PEGMEMA)–CH2–CH2 peak (δ = 0.8–1.2 ppm).
CH2, δ = 5.7–6.0 ppm), P(PEGMEMA) –CH2–CH2 peak (δ = 3.8–4.2 ppm) and block copolymer –CH2–CH2 peak (δ = 0.8–1.2 ppm).
Cell culture
The human ovarian cancer cell lines OVCAR-3 were grown in RPMI-1640 [2 × 10−3 M L-glutamine, 1.5 g L−1 sodium bicarbonate, 0.010 M 2-hydroxyethylpiperazinesulfonic acid (HEPES), 4.5 g L−1 glucose, 10−3 M sodium pyruvate] medium supplemented with 10% foetal bovine serum (FBS). The cells were grown in 5% CO2 at 37 °C.In vitro cell proliferation assay
OVCAR-3 cell lines were seeded in 96-well plates (3000 cells per well) with culture medium 10% FBS RPMI at 37 °C in 5% CO2 for 24 h. The medium was refreshed with 0.2 mL of a solution consisting of 0.1 mL medium and 0.1 mL of micellar solution of PMMA–b-P(PEGMEMA) block copolymer micelles to reach a final micelle concentration of 10, 25, 50, 125, 250, 500 and 1000 μg mL−1 followed by incubation at 37 °C in the incubator for 72 h. Subsequently, the medium was removed and the cells were fixed in 10% (w/v) TCA for 30 min at 4 °C and washed 5 times with tap water followed by staining with 0.4% (w/v) SRB dissolved in 1% acetic acid for 20 min and washed again 5 times with 1% acetic acid. After drying overnight, 100 μL of 0.010 M Tris (pH = 10.5) solution was added to solubilize the dye. Absorbance was measured at 570 nm using a S960 plate reader (Metertech, Taiwan). Non-treated cells were used as controls. The optical density (OD) was used to calculate cell viability.| Cell viability (%) = (OD570, sample − OD570, blank)/(OD570, control − OD570, blank) × 100 |
The experiments were carried out using six replicas.
OVCAR-3 cells were incubated in 24-well plates (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well) with culture medium 10% FBS RPMI-1640 at 37 °C in 5% CO2 for 24 h. The medium was refreshed and 0.5 mL solution was added to each well, which was prepared from 0.25 mL 10% FBS RPMI-1640 and 0.25 mL distilled water with Nile Red or fluorescent micelles, leading to final concentrations of 10 μg mL−1 Nile Red and 100 μg mL−1 micelles. The solutions were subsequently incubated at 37 °C in 5% CO2 for 30 min and 2 h. The OVCAR-3 cells were washed 2 times with PBS solution.
000 cells per well) with culture medium 10% FBS RPMI-1640 at 37 °C in 5% CO2 for 24 h. The medium was refreshed and 0.5 mL solution was added to each well, which was prepared from 0.25 mL 10% FBS RPMI-1640 and 0.25 mL distilled water with Nile Red or fluorescent micelles, leading to final concentrations of 10 μg mL−1 Nile Red and 100 μg mL−1 micelles. The solutions were subsequently incubated at 37 °C in 5% CO2 for 30 min and 2 h. The OVCAR-3 cells were washed 2 times with PBS solution.
Quantitative analysis. The amount of micelles or dye taken up was quantified by measuring the fluorescence intensity of the solution in the medium after different time intervals. The fluorescence intensity of the medium was measured at λex = 460 nm and λem = 512 nm for the micelles and λex = 530 nm and λem = 625 nm for Nile Red. The values were then compared to the fluorescence intensity of the solution at time 0. The experiments were carried out in triplicates. The standard deviation was calculated from three independent measurements.
Laser scanning confocal microscopy. Micrographs were recorded on live cells after incubation with the micelle or dye solution for a preset time.
In order to obtain a three-dimensional picture with stained nucleus and lysosomes, the OVCAR-3 cells were seeded in a 35 mm Fluorodish (World Precision Instruments) and incubated for 2 days. A solution of micelles (100 μg mL−1) was added to the OVCAR-3 cells and the mixture was incubated at 37 °C for 2 h, similar to the procedure described above. The cells were washed thrice with PBS and subsequently stained with 2 μg mL−1 Hoechst 33342 (Invitrogen) for 5 min, followed by two washing steps with PBS. The cells were then stained with 100 nM LysoTracker Red DND-99 (Invitrogen) for 1 min. The dye solution was quickly removed and the cells were gently washed with PBS. Finally, the cells were mounted in PBS and observed under a laser scanning confocal microscope (Zeiss LSM 780).
000 cells per well) with culture medium 10% FBS RPMI-1640 at 37 °C in 5% CO2 for 24 h. Cells were pre-incubated for 30 min at 37 °C with various endocytosis inhibitors diluted in the RPMI media: sodium azide (2.3 mM), 2-hydroxypropyl-β-cyclodextrin (50 mM) and hypertonic sucrose (0.45 M) solution. The medium was refreshed and 0.5 mL solution was added to each well, which was prepared from 0.25 mL of 10% FBS RPMI-1640 and 0.25 mL of the micellar solution (final micelle concentration 100 μg mL−1). The micellar solutions were incubated for 30 min and 2 h with the same concentrations of inhibitors present during subsequent incubation with the copolymer. The mean fluorescence intensity was obtained using the same settings and methods described above. The experiments were carried out in triplicates. The standard deviation was calculated from three independent measurements.
Analysis
Results and discussion
Synthesis
The RAFT polymerization technique has been commonly used as a robust method to synthesize polymers with complex architectures including block copolymers.8,36–38 The RAFT process is suitable for a range of monomer types and is resilient to the presence of many functional groups.5The RAFT agent 4-cyanopentanoic acid dithiobenzoate (CPADB) was employed in the polymerization of MMA in toluene at 70 °C to obtain the PMMA macroRAFT agent. The conversion of the monomer was determined via NMR spectroscopy comparing the intensities of the vinyl proton peaks (5.6 and 6.1 ppm) to the aliphatic proton peaks (1.1–1.4 ppm). After 24 h of reaction time, PMMA with a theoretical molecular weight of 4665 g mol−1 (SEC = 5000 g mol−1) and a very narrow molecular weight distribution with a PDI (polydispersity index) of 1.06 was obtained. The resulting PMMA homopolymer was used as a macroRAFT agent in the polymerization of PEGMEMA (Scheme 1) in toluene at 70 °C leading to PMMA47-b-P(PEGMEMA)46 block copolymers. The theoretical molecular weight was calculated to be 18500 g mol−1 based on 1H NMR and the experimental value (SEC) was 33000 g mol−1 with a PDI of 1.16. The deviation of the theoretical and experimental molecular weights (homopolymer and block copolymer) can be assigned to the polystyrene SEC calibrations. The SEC profile (ESI, Fig. S1†) reveals the shift of the polymer peaks to higher molecular weights after chain extension. To avoid PEGMEMA homopolymerization, the concentration of the initiator was kept below the macroRAFT agent concentration.38 In a similar manner PMMA47-b-P(PEGMEMA45-co-MAA10) was prepared, which has a theoretical molecular weight of 19300 g mol−1 and an experimental molecular weight (SEC) of 46000 g mol−1 (ESI, Fig. S2†).
Micelles were prepared by dissolving the copolymers in a common organic solvent, here DMF, followed by dialysis against distilled water resulting in the formation of micelles with PMMA in the core and P(PEGMEMA) or P(PEGMEMA-co-MAA) in the shell of the micelle. Dynamic light scattering (DLS) and transmission electron microscopy (TEM) were used to determine the shape and size of the micelles. Based on DLS, the hydrodynamic diameter of PMMA-b-P(PEGMEMA) was found to be about 20 nm with a narrow distribution and a PDI below 0.2 (ESI, Fig. S3†). The size of approximately 20 nm was confirmed using TEM (Fig. 1). The self-assembly of PMMA-b-P(PEGMEMA-co-MAA) resulted in the formation of micelles of 40 nm. Although the size of the block copolymer was similar to the one described above, the negatively charged MAA groups probably led to the stretching of the polymers chains in the shell, thus larger hydrodynamic diameters. The negative zeta-potential of ζ = −15 mV in water, which is slightly less than the micelle in the absence of PMAA groups ζ = −5 mV, confirms the presence of negative charges. The micelles were subsequently crosslinked using 1,8-diaminooctane according to an established procedure.33,39 Crosslinking was confirmed by the stability of the micelles in solvents such as DMF, which is a good solvent for both blocks (ESI, Fig. S4†). Both, SEC (ESI, Fig. S2†) and DLS (ESI, Fig. S4†) analysis, provide conclusive results for the successful crosslinking. The hydrodynamic diameter is now reduced to 25 nm due to the contractive forces of the crosslinking process as well as the loss of negative charges in the shell. An increase of the zeta potential ζ to +5 mV hints to a very small amount of free amine functionalities.
 | ||
| Fig. 1 Transmission electron microscope image of PMMA47-b-P(PEGMEMA)46 micelles after phosphotungstic acid negative staining. Bar = 100 nm. | ||
Cytotoxicity and endocytosis pathways of micelles
Human ovarian cancer cell lines (OVCAR-3) were chosen to test the interaction of micelles with tumour cells. The OVCAR-3 cell line is an attractive model because these cells are resistant to clinically relevant concentrations of many anti-cancer drugs and therefore may be used to study the mechanisms of drug resistance in ovarian cancer.40 A sulforhodamine B (SRB) assay, which was described earlier,41 was used to determine the cytotoxicity of the nanoparticles. Fig. 2 demonstrates the percentage viability of the cancer cells relative to the control after 72 h of incubation at 37 °C with polymer concentrations varying from 10 to 1000 μg mL−1. No significant levels of toxicity were recorded and concerns regarding the presence of the RAFT end functionality can be omitted.30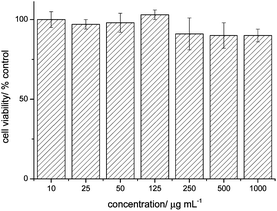 | ||
| Fig. 2 Cell viability percentage of OVCAR-3 cells after 72 h of incubation with PMMA47-b-P(PEGMEMA)46 micelles; n = 6. | ||
Ideally, the drug is taken up by a cell solely as cargo inside the micelle. This would ensure that the drug is mainly taken up via endocytosis by-passing the drug resistance mechanism. So far, it is commonly accepted that the cellular uptake of nanoparticles is dependent on a range of parameters; among them nanoparticle size, concentration and surface charge.23,42,43 Although the uptake of polymer micelles is the centre of attention of some selected publications, the mechanism is still not fully understood. In addition, the effect of crosslinking of micelles on cellular uptake is so far unknown. Since the aim is to deliver drug loaded micelles, we chose Nile Red and later Lucifer Yellow as model drugs for the exocytosis studies since they are both fluorescent and non-toxic. The drug carrier was labelled with a green fluorophore by adding 1 wt% of fluorescein acrylate to the polymerization of PEGMEMA and PEGMEMA/MAA, respectively.
Initial testing for endocytosis was carried out by inhibiting the energy-consuming endocytotic pathway by either lowering the temperature or by adding NaN3, a mitochondrial activity inhibitor that leads to the energy depletion of cells.44 Diffusion, in contrast, is not affected by these measures. A known number of cells (15000) were pre-treated for 30 min at 4 °C or with 2.3 mM of NaN3, followed by the addition of either micelles (100 μg mL−1 of crosslinked or non-crosslinked micelles) or Nile Red (10 μg mL−1). The cells were then incubated for a further 30 min or 2 h under these conditions. The amount of drug carrier or Nile Red taken up was quantified using the fluorescence intensity of the cell growth media. This intensity was compared to the fluorescence of the solution at time zero (Fig. 3 and 5).
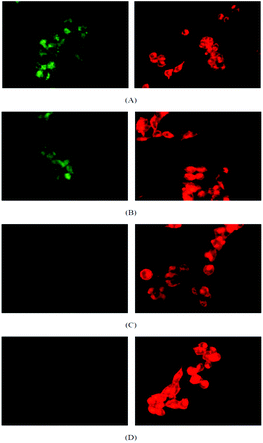 | ||
| Fig. 3 Confocal microscopy images OVCAR-3 cells after incubation with micelles (green) and Nile Red (red) at (A) 37 °C for 30 min, (B) 37 °C for 2 h, (C) 4 °C for 30 min and (D) 4 °C for 2 h. The amount of micelle (left column) inside the cell increases with increasing reaction time (A, B) while no uptake is observed at 4 °C (C, D). The uptake of Nile Red (right column) is independent from the temperature. | ||
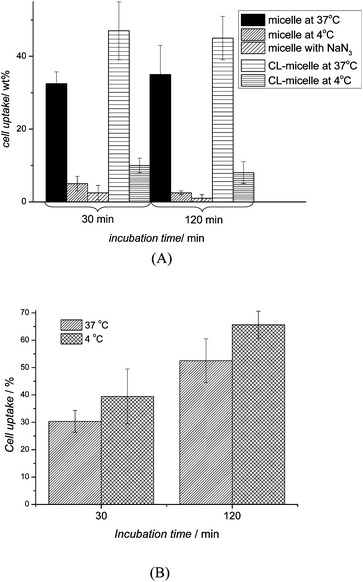 | ||
| Fig. 5 Cellular uptake percentage in OVCAR-3 cells after 30 and 120 min of (A) micelles (crosslinked (CL) and non-crosslinked) pre-treated at 37 °C, 4 °C and NaN3 and (B) Nile Red pre-treated at 37 °C and 4 °C (n = 3). The uptake of micelles and crosslinked micelles were inhibited at low temperatures and in the presence of NaN3 indicating uptake via endocytosis. In contrast, no effect was observed with Nile Red. | ||
Fig. 3 shows the confocal micrographs of live OVCAR-3 cells when incubated with micelles (100 μg mL−1) or Nile Red (10 μg mL−1) for 30 min or 2 h under conditions that inhibit endocytosis. Nile Red clearly diffused into the cells while the cellular uptake of polymeric micelles has been reduced considerably at low temperatures. The first indication that micelles were taken up by endocytosis can be detected in Fig. 3A. After 30 min, the green fluorescence of the micelles appeared localized in small spots, which might indicate that the micelles are concentrated within the lysosomes. To confirm that the micelles were indeed localized in the lysosomes, imaging of the live cells was carried out using a confocal fluorescent microscope (Fig. 4). The nucleus was stained in blue while the lysosomes were stained with a red lysotracker. Overlaying the pictures of the green fluorescent micelles and the red lysosomes confirms that both, micelles and lysosomes, are co-located (yellow). The 2D-pictures in two different magnifications can be found in the ESI (Fig. S5†).
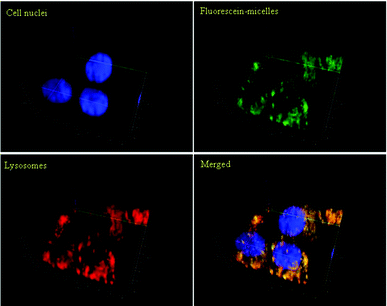 | ||
| Fig. 4 Confocal microphotographs of OVCAR-3 cells after incubation with micelles at 37 °C for 2 h. Polymeric micelles (green) were labelled with fluorescein. Cell nuclei (blue) were stained with Hoechst 33342; lysosomes (red) were stained with LysoTracker Red DND-99. The 3D images were generated using Zen 2011 (Zeiss) software. The yellow colour in the merged picture indicates that the location of the micelles is equivalent to the location of the lysosomes. | ||
Around 30 wt% of available micelles were taken up within 30 min. Extending the incubation period did not lead to a higher uptake indicating that the process is efficient and complete in less than an hour (Fig. 5A). While this fast uptake is not unusual, it needs to be considered that a fast uptake may also mean that the particles could easily be taken up by non-cancerous cell lines, which may lead to fast clearance or accumulation in the body. Further in vivo work is necessary using cell lines such as human monocytes, murine macrophages, non-parenchymal liver cells, and neutrophilic granulocytes to evaluate the suitability of using comb-type PEG instead of high-molecular weight PEG.45 At this point in time, the focus is on the type of uptake of these particles. Treating the cells with NaN3 or lowering the temperature to 4 °C led to a noteworthy drop in cellular uptake suggesting that the micelles enter the cells indeed via endocytosis. The uptake of crosslinked micelles is noticeably increased with more than 40 wt% being taken up within 30 min. Again, the uptake is reduced to less than 10 wt% in the presence of inhibitory factors indicating that the uptake of both, crosslinked and non-crosslinked micelles, proceeds along the same pathway. In contrast, small molecules such as Nile Red or drugs are unlikely to enter the cell by endocytosis with diffusion though the cell membrane being the main mechanism.42,46 Nile Red (M = 320 g mol−1) was therefore unaffected by conditions that might affect endocytosis such as low temperatures (Fig. 5B).
Cellular uptake of a particulate system can occur through various mechanisms such as phagocytosis, fluid phase pinocytosis, or endocytosis.47,48 Macrophages employ mainly phagocytosis to internalize particulate matter.47 However, as the lower size cut-off for phagocytosis is around 500 nm, this possibility was not considered as the nanoparticles here are ∼25 nm.49 Among the possible endocytosis pathways, clathrin mediated endocytosis and caveolae mediated endocytosis are the most commonly used routes for particles of this size.23 Clathrin mediated endocytosis is a continuous process for virus entry, which is usually rapid and efficient.27 It is known that positively charged macromolecules can be effectively internalized via clathrin mediated endocytosis after binding to the cell membrane's anionic sites such as glycosaminoglycans, asialoglycoprotein or heparan sulphate receptors.50 Hypertonic (0.4–0.5 M) sucrose represents one of the most popular chemical inhibitors of clathrin mediated endocytosis and it acts by blocking protein internalization.51 Therefore, OVCAR-3 cells were pre-treated with a hypertonic sucrose (0.5 M) solution for 30 min. The solutions containing either crosslinked or non-crosslinked micelles (100 μg mL−1) were then incubated with the treated cells and the cellular uptake was measured after 30 min and 2 h (Fig. 6). The weight percentage of endocytosed micelles did not deviate significantly from the control indicating that those polymeric micelles might not enter the cells via clathrin mediated endocytosis. Shell-crosslinked micelles in contrast seem to enter partly via clathrin-mediated endocytosis, although the fraction is not significant and may even be subject to error.
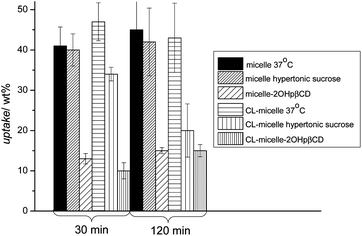 | ||
| Fig. 6 Effect of endocytosis inhibitors on the uptake of PPEGMEMA-b-PMMA micelles in OVCAR-3 cell after 30 and 120 min of incubation. Cell were pre-treated for 30 min with the corresponding inhibitors (no treatment for control) and incubated with the nanoparticles. The reduction of the micelle uptake in the presence of 2-hydroxypropyl-β-cyclodextrins points to caveolae mediated endocytosis of these micelles while inhibition of the clathrin mediated pathway with sucrose does not affect the amount of micelles taken up. | ||
Membrane (lipid) rafts and caveolae are cellular domains that play a vital role in cell function by concentrating plasma membrane proteins and lipids.52 The majority of pharmacological inhibitors of caveolae/lipid raft mediated endocytosis target cholesterol.53 There are several methods to inhibit this pathway such as inhibition of cholesterol biosynthesis by statins, cholesterol extraction from the plasma membrane using cyclodextrins, cholesterol sequestration within the membrane using polyene antibiotics such as nystatin or enzymatic modification of plasma membrane cholesterol by cholesterol oxidase.54 Here, cholesterol extraction using β-cyclodextrins was chosen. Methyl-β-cyclodextrin (MβCD) is the most common inhibitor used that alters the structure of cholesterol rich domains in cell membranes.55 Previous work by Rothblat et al. has demonstrated that 2-hydroxypropyl-β-cyclodextrin (2OHpβCD) removes cholesterol very efficiently from cell membranes, but it is considerably less toxic than the more widely used MβCD.56 Therefore, to inhibit caveolae mediated endocytosis, 15000 OVCAR-3 cells were pre-treated with 2OHpβCD (50 mM) for 30 min and incubated with the micellar solution with a polymer concentration of 100 μg mL−1 in the presence of 2OHpβCD. The effect of caveolae endocytosis inhibition on the uptake of micelles is shown in Fig. 6. It was found that the inhibition of caveolae mediated endocytosis reduces considerably the cellular uptake of micelles leading to the conclusion that caveolae mediated endocytosis is the main pathway. This is in contrast to the work by Sahay et al. who observed that Pluronic micelles were internalized by MDCK cells through clathrin-mediated endocytosis.23 This was explained with the possible interaction of the polymer with the cell membrane. Interestingly, below the CMC, the non-associated Pluronic macromolecules entered the cell via caveolae mediated endocytosis.23
Interesting is the comparison to crosslinked micelles. Within errors, the effect is similar and caveolae mediated endocytosis is again the preferred pathway. This is remarkable considering that the crosslinking of the shell of the micelle will partly limit the mobility of the water-soluble PPEGMEMA and fully prevent the interaction of the amphiphilic polymer with the cell membrane as is known for Pluronic.
At this point it needs to be considered that the micelle size is subject to a size distribution (Fig. S3 and S4†). Different sizes may behave slightly differently and micelles with sizes below 20 nm may even fall below the cut-off for endocytosis.57
Exocytosis and intracellular retention
The long-lasting effect of polymer–drug conjugates in clonogenic assays indicates that the success of a drug carrier might lie in the retention of a drug carrier within the cell preventing early exocytosis.29 Exocytosis is the process where vesicles merge with the plasma membrane to release the content from the cell. Lipid rafts and more particularly cholesterol are involved in the regulation of exocytosis.58 For example, it has been reported that about 110000 endocytic vesicles of about 100 nm in diameter are formed per cell per hour resulting in 20% of the plasma membrane being recycled per hour in rat adipocytes.59 The intracellular level of nanoparticles increases according to the concentration of nanoparticles in the extracellular medium although the increase is not linear with increasing nanoparticle concentration.8 This equilibrium is disturbed when the nanoparticle suspension was replaced with fresh medium resulting in a drop in extracellular nanoparticle level. It has been shown that at least two intracellular compartments could be involved during exocytosis: the first which turns over rapidly, and the second, which turns over relatively slowly.60 Small molecules or drugs can be recycled through the fast turnover compartment whereas nanoparticles with a certain size were rather transported in the second slow turnover compartment.61,62
OVCAR-3 cells were therefore incubated with fluorescently labelled micelles (crosslinked or uncrosslinked) or with Lucifer yellow. Lucifer yellow was used instead of Nile Red since its hydrophilic nature makes it a fluid marker and allows better release while Nile Red tends to adhere to the cell. After the equilibrium was established, the cells were washed with fresh media and the release of Lucifer yellow and the two types of micelles was monitored. It was observed that about 40% of the fluid phase marker leached from the cells within 2 h while only 4% of the micelles exocytosed into the fresh media (Fig. 7). This result might be due to the Lucifer yellow being trapped at the level of the rapid recycling compartment and therefore, is available for immediate excretion from the cells. As mentioned earlier, the difference in the exocytosis rate between the micelles and the fluid phase marker could be attributed to the difference in their nature and size. It has been reported that the size of the molecules affects the transfer from the fast recycling compartment to the slow recycling compartment as well as the rate of recycling from the slow recycling compartment.63,64 For example, it was previously demonstrated that Lucifer yellow was trapped within the rapid recycling compartments63 while the large molecule horseradish peroxidase was located in the slow recycling compartment, hence leading to a slow rate of exocytosis.64Fig. 8 shows visual results of the exocytosis of Nile Red (replacing Lucifer yellow) and micelles after 10, 30, 60 and 120 min of cell washing and addition of fresh media. The dye leached immediately into the media as evidenced by the red halo around the cells. The cells were washed and replaced with fresh media leading to a decline in fluorescence intensity inside the cells over time. In contrast, the uncrosslinked micelles did not show a decline in fluorescence intensity over time despite the washing steps at 10, 30, 60 and 120 min. It seems therefore that the exit of the micelles from the cells is delayed. The visual results from Fig. 8 showed good correlation with the numerical results obtained in Fig. 7.
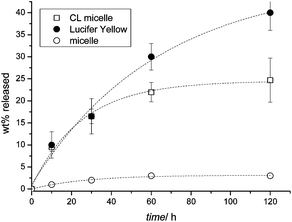 | ||
| Fig. 7 Release kinetics of micelles and Lucifer yellow for OVCAR-3 cells. Cells were incubated at 37 °C with micelles and Lucifer yellow for 30 min, then washed twice with PBS and the medium replaced with a new medium. The fluorescence intensity of the cell growth medium was directly correlated to the amount of dye or micelle that was released. | ||
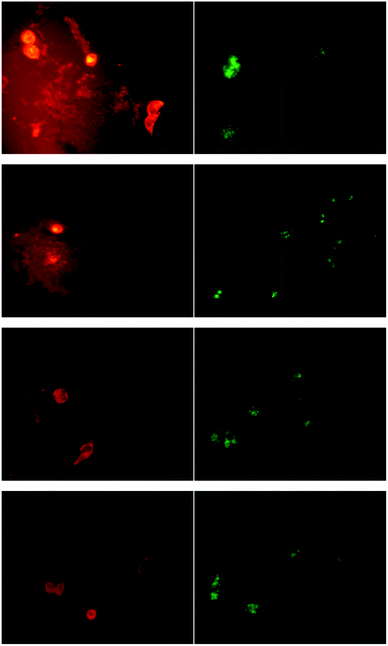 | ||
| Fig. 8 Confocal fluorescent micrographs of live OVCAR-3 cells showing the release of Nile Red (red) and micelles (green). The cells were incubated with the dye or micelles, washed, and then placed in fresh media. The release was then monitored after 10, 30, 60 and 120 min (from top to bottom), following by replacement of the cell growth media. Nile Red was released into the media very quickly (red halo around the cells) and after 120 min the reduced fluorescence intensity indicates that Nile Red has been released. The fluorescence intensity of the micelles does not change noticeably indicating a very slow release. | ||
Interestingly, the behaviour of crosslinked micelles deviates noticeably from that of the uncrosslinked one. The rate of exocytosis is close to the rate of Lucifer yellow (although direct comparison with the dye is not appropriate). This result needs to be seen under the light that the amount of micelles, crosslinked and non-crosslinked, inside the cells was almost identical (Fig. 5). The fast exocytosis of the crosslinked micelles is similar to that of hard nanoparticles such as nanotubes,65 where exocytosis is similar in rate to endocytosis. The difference of both nanoparticles in terms of exocytosis may be their stability. Crosslinked micelles are unaffected by any environmental changes and they remain stable throughout the whole process. Micelles in contrast are in theory dynamic structures that can disassemble upon concentration changes, but also in the presence of other molecules that can interact with the micelle.33 In fact, the intracellular matrix is rather crowded with macromolecules that can interact with micelles.66 The disassembly of micelles might not allow exocytosis, rather the single amphiphilic block copolymers may start interacting with the cell membrane.42 It should be noted here that although micelles could potentially disassemble, they usually have a high kinetic stability. Especially micelles with a core made from a polymer with a high glass transition temperature Tg—as is the case here—should be indefinitely stable against disassembly. Curiously, we have found that at very low concentrations, below the cmc, cross-linked micelles behave differently to the ones that are uncrosslinked.33 This behaviour is independent from the glass transition temperature and the effect is visible in high Tg and low Tg micelles resulting in an increased toxicity of drug loaded cross-linked micelles.34 The precise origin of this observation is unknown and warrants further investigation. Earlier studies also showed only a slight depression of the glass transition temperature of PMMA by 5 °C, which was assigned to incomplete phase-separation with P(PEGMEMA) and the confinement of PMMA into a nano-space.34 This depression is not sufficient to argue for the decreased stability. One reason however could be that the molecular weight of the PMMA used in this investigation is comparatively low compared to the P(PEGMEMA), which allows less entanglements leading to a lower kinetic stability.
Exocytosis was studied in more detail by inhibiting the process using Filipin III (25 μg mL−1). The inhibition process was performed in serum free medium as it has been reported that the presence of bovine serum albumin (BSA) or growth factors such as PDGF, restores the process completely and even induces exocytosis.61 Thus, OVCAR-3 cells were pre-treated with Filipin for 30 min. Then, micelles and Lucifer yellow (100 μg mL−1) were incubated with the cell lines in the presence of the inhibitor for 1 h. Cells were washed with PBS twice to remove micelles and Lucifer yellow in the extracellular milieu. Fresh cell growth medium containing the inhibitor was added and the fluorescence intensity after 30 and 120 min was measured. Inhibition of the exocytosis process led to the further decrease of an already very slow process (Fig. 9(A)) indicating that the main pathway out of the cell was indeed by exocytosis. In contrast, it could be observed that a large amount of Lucifer yellow was partly externalized from the cells even when the exocytosis process was inhibited (Fig. 9(B)) suggesting that other transport pathways are active such as organic anion transport.67
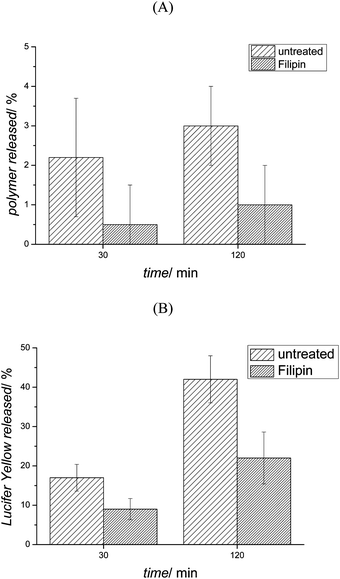 | ||
| Fig. 9 Filipin effect on (A) micelle and (B) Lucifer yellow exocytosis. OVCAR-3 cells were pre-treated 30 min with Filipin (no pre-treatment for the control) and incubated with micelles and Lucifer yellow for 1 h, then washed twice with PBS and new medium was replaced. The fluorescence intensity of the medium was measured after 30 and 120 min, respectively. | ||
Conclusion
Understanding the mode of entry of a nanocarrier into the cell is a vital process, especially when trying to by-pass the drug resistance mechanism of the cell. Micelles are widely proposed as drug carriers, but recently more and more publications propose the use of crosslinked micelles to stabilize the inherent dynamic structure of the micelle. It has frequently been observed that crosslinking affects drug loading and release as well as the amount of uptake by the cell. To date, it is unknown what effect crosslinking might have on the mode of entry of the drug carrier into the cell and the length of stay inside the cell, which may have detrimental consequences on the delivery of the drug. This work was inspired by the observation that tumour cells continued to die after treatment with drug loaded micelles although the cells were washed with fresh media. The drug-loaded carrier must have been entrapped inside the cell, where it continued affecting the cells. Here, we investigated the endocytosis and exocytosis of micelles and shell-crosslinked micelles in more detail. Interestingly, there was no significant effect of shell-crosslinking on the pathway of endocytosis with caveolae mediated endocytosis being the main type of entry. The difference between both carriers became obvious when quantifying the exocytosis of the carrier. The uncrosslinked micelles were trapped inside the cell and almost no release was measured after 2 hours. Crosslinking of the micelle in contrast led to fast exocytosis (25% in 2 hours). This could have significant implications on the activity of the drug carrier, especially when cleavage of the drug from the polymer is required. However one can argue that the fast endo- and exocytosis of crosslinked micelles may be beneficial when transport across the cell is envisaged.Acknowledgements
MS thanks the ARC (Australian Research Council) for funding in the form of a Future Fellowship (FT0991273). MS, MP and DM thank the ARC for financial support for this project (DP110102409). The authors would like to thank the Centre for Advanced Macromolecular Design (CAMD) and UNSW Mark Wainwright Analytical Centre for support.Notes and references
- R. Savić, A. Eisenberg and D. Maysinger, J. Drug Targeting, 2006, 14, 343–355 CrossRef CAS.
- L. E. van Vlerken and M. M. Amiji, Expert Opin. Drug Delivery, 2006, 3, 205–216 Search PubMed.
- A. T. Florence, J. Controlled Release, 2012, 164, 115–124 Search PubMed.
- Y. Yan, C. J. Ochs, G. K. Such, J. K. Heath, E. C. Nice and F. Caruso, Adv. Mater., 2010, 22, 5398–5403 CrossRef CAS.
- M. H. Stenzel, Chem. Commun., 2008, 3486–3503 RSC.
- K. E. Uhrich, S. M. Cannizzaro, R. S. Langer and K. M. Shakesheff, Chem. Rev., 1999, 99, 3181–3198 CrossRef CAS.
- J. Liu, Y. Xiao and C. Allen, J. Pharm. Sci., 2004, 93, 132–143 CrossRef CAS.
- Y. Kim, M. H. Pourgholami, D. L. Morris and M. H. Stenzel, Macromol. Biosci., 2011, 11, 219–233 Search PubMed.
- T. Hamaguchi, Y. Matsumura, M. Suzuki, K. Shimizu, R. Goda, I. Nakamura, I. Nakatomi, M. Yokoyama, K. Kataoka and T. Kakizoe, Br. J. Cancer, 2005, 92, 1240–1246 CrossRef CAS.
- Y. Kim, M. H. Pourgholami, D. L. Morris and M. H. Stenzel, J. Mater. Chem., 2011, 21, 12777–12783 RSC.
- E. V. Batrakova, S. Li, S. V. Vinogradov, V. Y. Alakhov, D. W. Miller and A. V. Kabanov, J Pharmacol. Exp. Ther., 2001, 299, 483–493 Search PubMed.
- N. Rapoport, A. Marin, Y. Luo, G. D. Prestwich and M. Muniruzzaman, J. Pharm. Sci., 2002, 91, 157–170 CrossRef CAS.
- M. Muniruzzaman, A. Marin, Y. Luo, G. D. Prestwich, W. G. Pitt, G. Husseini and N. Y. Rapoport, Colloids Surf., B, 2002, 25, 233–241 CrossRef CAS.
- T. Demina, I. Grozdova, O. Krylova, A. Zhirnov, V. Istratov, H. Frey, H. Kautz and N. Melik-Nubarov, Biochemistry, 2005, 44, 4042–4054 CrossRef CAS.
- J. Akimoto, M. Nakayama, K. Sakai and T. Okano, Mol. Pharmaceutics, 2010, 7, 926–935 CAS.
- H. Z. Zhao and L. Y. L. Yung, J. Biomed. Mater. Res., Part A, 2009, 91A, 505–518 Search PubMed.
- H. C. Tsai, W. H. Chang, C. L. Lo, C. H. Tsai, C. H. Chang, T. W. Ou, T. C. Yen and G. H. Hsiue, Biomaterials, 2010, 31, 2293–2301 CrossRef.
- P. A. Ma, S. Liu, Y. B. Huang, X. S. Chen, L. P. Zhang and X. B. Jing, Biomaterials, 2010, 31, 2646–2654 CrossRef CAS.
- X. B. Xiong, H. Uludag and A. Lavasanifar, Biomaterials, 2010, 31, 5886–5893 Search PubMed.
- F. Q. Hu, X. H. Jiang, X. Huang, X. L. Wu, H. Yuan, X. H. Wei and Y. Z. Du, J. Drug Targeting, 2009, 17, 384–391 CrossRef CAS.
- A. Verma and F. Stellacci, Small, 2010, 6, 12–21 CrossRef CAS.
- S. D. Conner and S. L. Schmid, Nature, 2003, 422, 37–44 CrossRef CAS.
- G. Sahay, E. V. Batrakova and A. V. Kabanov, Bioconjugate Chem., 2008, 19, 2023–2029 CrossRef CAS.
- K. R. Peters, W. W. Carley and G. E. Palade, J. Cell Biol., 1985, 101, 2233–2238 Search PubMed.
- K. G. Rothberg, J. E. Heuser, W. C. Donzell, Y.-S. Ying, J. R. Glenney and R. G. W. Anderson, Cell, 1992, 68, 673–682 CAS.
- L. A. Carver and J. E. Schnitzer, Nat. Rev. Cancer, 2003, 3, 571–581 CrossRef CAS.
- H. Hillaireau and P. Couvreur, Cell. Mol. Life Sci., 2009, 66, 2873–2896 CrossRef CAS.
- H. Y. Nam, S. M. Kwon, H. Chung, S.-Y. Lee, S.-H. Kwon, H. Jeon, Y. Kim, J. H. Park, J. Kim, S. Her, Y.-K. Oh, I. C. Kwon, K. Kim and S. Y. Jeong, J. Controlled Release, 2009, 135, 259–267 CrossRef CAS.
- V. T. Huynh, J. Y. Quek, P. L. de Souza and M. H. Stenzel, Biomacromolecules, 2012, 13, 1010–1023 CrossRef CAS.
- A. Gregory and M. H. Stenzel, Expert Opin. Drug Delivery, 2011, 8, 237–269 Search PubMed.
- C. F. van Nostrum, Soft Matter, 2011, 7, 3246–3259 RSC.
- A. M. Nyström and K. L. Wooley, Acc. Chem. Res., 2011, 44, 969–978 CrossRef CAS.
- Y. Kim, M. H. Pourgholami, D. L. Morris and M. H. Stenzel, Biomacromolecules, 2012, 13, 814–825 CrossRef CAS.
- Y. Kim, E. D. Liemmawal, M. H. Pourgholami, D. L. Morris and M. H. Stenzel, Macromolecules, 2012, 45, 5451–5462 Search PubMed.
- Y. Mitsukami, M. S. Donovan, A. B. Lowe and C. L. McCormick, Macromolecules, 2001, 34, 2248–2256 CrossRef CAS.
- G. Moad, R. T. A. Mayadunne, E. Rizzardo, M. Skidmore and S. H. Thang, Macromol. Symp., 2003, 192, 1–12 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- A. Gregory and M. H. Stenzel, Prog. Polym. Sci., 2012, 37, 38–105 CrossRef CAS.
- Q. Zhang, E. E. Remsen and K. L. Wooley, J. Am. Chem. Soc., 2000, 122, 3642–3651 CrossRef CAS.
- T. C. Hamilton, R. C. Young, W. M. McKoy, K. R. Grotzinger, J. A. Green, E. W. Chu, J. Whang-Peng, A. M. Rogan, W. R. Green and R. F. Ozols, Cancer Res., 1983, 43, 5379–5389 Search PubMed.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS.
- J. Zastre, J. Jackson, M. Bajwa, R. Liggins, F. Iqbal and H. Burt, Eur. J. Pharm. Biopharm., 2002, 54, 299–309 CrossRef CAS.
- M. Liang, I. C. Lin, M. R. Whittaker, R. F. Minchin, M. J. Monteiro and I. Toth, ACS Nano, 2009, 4, 403–413.
- C. Y. Dombu, M. Kroubi, R. Zibouche, R. Matran and D. Betbeder, Nanotechnology, 2010, 21, 355102 Search PubMed.
- D. E. Owens Iii and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef CAS.
- E. V. Batrakova, H.-Y. Han, V. Y. Alakhov, D. W. Miller and A. V. Kabanov, Pharm. Res., 1998, 15, 850–855 Search PubMed.
- K. A. Foster, M. Yazdanian and K. L. Audus, J. Pharm. Pharmacol., 2001, 53, 57–66 Search PubMed.
- H. Suh, B. Jeong, F. Liu and S. W. Kim, Pharm. Res., 1998, 15, 1495–1498 CAS.
- A. Rupper and J. Cardelli, Biochim. Biophys. Acta, Gen. Subj., 2001, 1525, 205–216 CrossRef CAS.
- G. Poon and J. Gariepy, Biochem. Soc. Trans., 2007, 35, 788–793 CrossRef CAS.
- J. Inal, S. Miot and J. A. Schifferli, Exp. Cell Res., 2005, 310, 54–65 Search PubMed.
- K. Simons and D. Toomre, Nat. Rev. Mol. Cell Biol., 2000, 1, 31–39 CrossRef CAS.
- R. G. Parton and A. A. Richards, Traffic, 2003, 4, 724–738 CrossRef CAS.
- E. J. Smart and R. G. W. Anderson, in Methods in Enzymology, ed. L. P. Chandan and K. Sen, Academic Press, 2002, pp. 131–139 Search PubMed.
- T. Irie, K. Fukunaga and J. Pitha, J. Pharm. Sci., 1992, 81, 521–523 CrossRef CAS.
- G. H. Rothblat, M. de la Llera-Moya, V. Atger, G. Kellner-Weibel, D. L. Williams and M. C. Phillips, J. Lipid Res., 1999, 40, 781–796 Search PubMed.
- A. Chaudhuri, G. Battaglia and R. Golestanian, Phys. Biol., 2011, 8, 046002 Search PubMed.
- C. Salaün, D. J. James and L. H. Chamberlain, Traffic, 2004, 5, 255–264 CrossRef.
- E. M. Gibbs and G. E. Lienhard, J. Cell. Physiol., 1984, 121, 569–575 Search PubMed.
- B. Goud, C. Jouanne and J.-C. Antoine, Exp. Cell Res., 1984, 153, 218–235 Search PubMed.
- J. Panyam and V. Labhasetwar, Pharm. Res., 2003, 20, 212–220 CrossRef CAS.
- J. Gruenberg, Nat. Rev. Mol. Cell Biol., 2001, 2, 721–730 CrossRef CAS.
- J. A. Swanson, B. D. Yirinec and S. C. Silverstein, J. Cell Biol., 1985, 100, 851–859 Search PubMed.
- M. J. Buckmaster, D. Lo Braico Jr., A. L. Ferris and B. Storrie, Cell Biol. Int. Rep., 1987, 11, 501–507 Search PubMed.
- H. Jin, D. A. Heller and M. S. Strano, Nano Lett., 2008, 8, 1577–1585 CrossRef.
- R. J. Ellis, Trends Biochem. Sci., 2001, 26, 597–604 CrossRef CAS.
- T. H. Steinberg, J. A. Swanson and S. C. Silverstein, J. Cell Biol., 1988, 107, 887–896 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The supporting information contains the size exclusion chromatograms (SECs) of the polymers as well as the dynamic light scattering (DLS) results of the micelles and the confocal fluorescent images of OVCAR-3 cells incubated with micelles at two magnifications (stained nucleus and lysosome). See DOI: 10.1039/c2bm00096b |
| This journal is © The Royal Society of Chemistry 2013 |