 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent developments in electromembrane extraction
Astrid
Gjelstad
*a and
Stig
Pedersen-Bjergaard
ab
aDepartment of Pharmaceutical Chemistry, School of Pharmacy, University of Oslo, P. O. Box 1068 Blindern, 0316 Oslo, Norway. E-mail: astridgj@farmasi.uio.no; Fax: +47 22 85 44 02; Tel: +47 22 85 75 58
bDepartment of Pharmacy, Faculty of Health and Medical Sciences, University of Copenhagen, DK-2100 Copenhagen, Denmark
First published on 26th June 2013
Abstract
Electromembrane extraction (EME) was published for the first time in 2006, and is a liquid-phase microextraction technique intended for analytical sample preparation. In EME, charged analytes are extracted in an electrical field, from the aqueous sample solution, through a supported liquid membrane and into an aqueous acceptor phase. EME is still in an early stage of development, although nearly 80 research papers have been published on the subject. The current paper reviews the EME literature with focus on applications and technical development, and critically discusses the future of the technology.
 Astrid Gjelstad | Astrid Gjelstad holds an associate professor position with the School of Pharmacy, University of Oslo, Norway. The field of interest is development of new sampling and microextraction technology for rapid and selective isolation of drugs and peptides from small amounts of biological matrices. The microextraction technology is based on artificial liquid membranes where studies of the transport across the membranes forced by either passive diffusion (liquid-phase microextraction, LPME) or electrokinetic migration (electromembrane extraction, EME) are special focus areas. New materials for dried blood spot sampling are also of interest. |
 Stig Pedersen-Bjergaard | Stig Pedersen-Bjergaard is a professor at the School of Pharmacy, University of Oslo, Norway and professor II at the Department of Pharmacy, University of Copenhagen, Denmark. The main focus is directed towards the development of new and improved microextraction concepts and techniques for rapid and selective isolation of drug substances and peptides from small volumes of biological fluids. The microextraction techniques are based on artificial liquid membranes, and mass transfer is based primarily on passive diffusion and on electrokinetic migration. The objective is to use the microextraction techniques for drug analysis and biochemical analysis in the future for rapid sampling and sample preparation in microscale laboratory systems, in single cells, and in living organisms. |
Introduction
Electromembrane extraction (EME) is a liquid-phase microextraction technique and was first published in 2006.1 EME is intended for sample preparation prior to (but not limited to) techniques like liquid chromatography, capillary electrophoresis, and mass spectrometry. The basic principle of EME is illustrated in Fig. 1. Target analytes are extracted from an aqueous sample, through an organic solvent immobilized as a supported liquid membrane (SLM) in the pores in the wall of a porous hollow fibre, and finally into an aqueous acceptor phase located inside the lumen of the hollow fibre. After EME, the acceptor phase is collected and subjected to the final chemical measurement. The driving force for the extraction is an electrical potential (dc) sustained over the SLM, with one electrode located in the sample and the other electrode located in the acceptor phase. Thus, EME is intended for charged analytes, and involves electrokinetic migration of the analytes across the SLM. For extraction of basic analytes, the anode is placed in the sample and the cathode is placed in the acceptor phase. In addition, pH is controlled in both the sample and the acceptor to make sure that the analytes are positively charged. For extraction of acidic analytes, the direction of the electrical potential is reversed, and the pH conditions are adjusted to make sure that the analytes are negatively charged.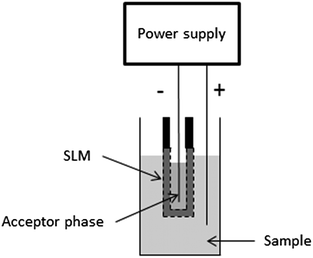 | ||
| Fig. 1 Principle of EME. | ||
Essentially, EME evolved from hollow fibre liquid-phase microextraction (LPME),2 where basic or acidic analytes are extracted from an aqueous sample, through an SLM and into an acceptor phase based on a pH gradient. Although LPME has received substantial attention in recent years,3 extraction times are relatively long due to slow mass transfer across the SLM. However, it was discovered that application of an electrical potential across the SLM increased the mass transfer significantly, and EME was introduced as a rapid alternative to LPME in 2006.1 Thus, by changing from LPME to EME, extraction times were typically reduced from 45 to 5 minutes.4 Major matrix constituents of biological and environmental samples are not able to pass the SLM, and are not extracted into the acceptor phase. Thus, in addition to rapid extraction, EME also gives excellent clean-up of biological5 and environmental samples6 (Fig. 2). Because target analytes are extracted into an aqueous acceptor phase in EME, the technique is directly compatible with high-performance liquid chromatography (HPLC), liquid chromatography-mass spectrometry (LC-MS), and capillary electrophoresis (CE). This will be discussed in more detail below, along with examples from extractions from different biological and environmental sample matrices.
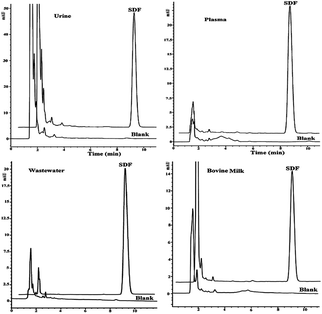 | ||
| Fig. 2 Examples of clean chromatograms obtained from EME of sodium diclofenac from different matrices followed by HPLC-UV. Reproduced with permission.33 | ||
Compared to existing extraction methods like liquid–liquid extraction (LLE) and solid-phase extraction (SPE), EME is remarkable in two different aspects. Firstly, EME can be conducted with extremely low consumption of organic solvents and consumables. Secondly, EME offers unique possibilities to adjust the extraction selectivity. The selectivity of the extraction can easily be controlled with the external power supply by the direction and the magnitude of the electrical potential. The direction controls if cations or anions are to be extracted, whereas the magnitude of the electrical potential controls the type of compounds to be extracted. Furthermore, the selectivity of the extraction can also be manipulated by the type of organic solvent used as the SLM. This will be discussed in further details below.
Although EME is still in a very early stage of development, nearly 80 papers have been published in recent years. This review gives an overview of the current status of EME, and also discusses the future perspectives for this alternative sample preparation technique.
Extraction kinetics
As discussed in the introduction, EME evolved from LPME based on the observation that mass transfer across the SLM was significantly promoted by an electrical potential.1 This finding has been confirmed in several recent publications, which have compared the extraction kinetics of LPME and EME.7–9 The kinetic aspects of EME are illustrated in Fig. 3 for the extraction of five different basic drug substances.10 As seen from curve C in Fig. 3, the sample is rapidly depleted for target analytes. Thus, the mass transfer from the sample and into the SLM is very efficient under EME conditions. Curve B in Fig. 3 illustrates the amount of analyte present in the SLM versus time, and as seen from this figure, also the mass transfer across the SLM is highly efficient. The residence time for the analyte in the SLM is relatively short, and for most of the drug substances the amount of analyte trapped in the SLM is low. The net effect of this is illustrated in curve A (Fig. 3), where the extracted amount of analyte is plotted versus extraction time. EME is rapid, and after 5 minutes of extraction, recoveries are between 60 and 90% and remain at this level even when extractions are performed for longer periods of time. In LPME on the other hand, the mass transfer across the SLM is relatively slow, and the recovery in the acceptor phase increases slowly versus time.9 Thus, the rate limiting step in LPME is the transfer of uncharged analytes across the SLM which is only by passive diffusion. In EME, there is a strong electro-kinetic transport of charged analytes across the SLM, and this is the principal reason for the rapid kinetics in EME.10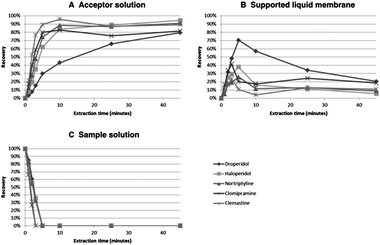 | ||
| Fig. 3 Distribution profiles for three phases of an EME system: the acceptor phase (A), SLM (B), and the sample (C). Acceptor and sample solution: 10 mM HCl, SLM: NPOE, voltage: 200 V, agitation: 900 rpm. Reproduced with permission.10 | ||
EME combined with different analysis methods
One of the advantages with electromembrane extraction (EME) is the direct compatibility with a wide range of analytical instruments. Extraction in the three-phase system, where the SLM is sandwiched between two aqueous solutions (the sample and the acceptor phase, respectively), provides an aqueous extract which can be directly injected into e.g. CE or HPLC.The first EME experiments were performed in combination with capillary electrophoresis with ultraviolet detection (CE-UV).1,11,12 Acceptor phases from EME containing small-molecule pharmaceuticals were separated and detected with CE-UV because of the speed, reduction in solvent consumption and disposal and the rapid method development offered by CE. Despite the relatively poor sensitivity, a lot of fundamental studies of EME regards to different compounds,11–15 extraction kinetics16 and studies of the SLM composition11 have been performed with CE.
Another advantage is the possibility for chiral separation in CE. This was exploited in three recent publications, where EME was combined with chiral separations.17–19 Nojavan et al. used EME for extraction of amlodipine enantiomers from plasma and urine samples followed by cyclodextrin (CD)-modified CE.17 This combination resulted in high enrichment and efficient sample cleanup, with limits of detection (LOD) in the lower ng mL−1 range. The obtained results showed that there was no discrimination between the two enantiomers in the extraction process. Similar results were obtained by the group of Fakhari and co-workers, which combined EME of trimipramine enantiomers18 with CD-modified CE. They concluded that the method had acceptable precision and was a highly sensitive technique for determination of trace amounts of trimipramine enantiomers in plasma and urine samples. EME of phenoxy acid herbicides followed by CD-modified CE are also described.20 The same group reported recently the combination of EME with maltodextrin (MD)-modified CE for separation, preconcentration and determination of tolterodine enantiomers in biological fluids.19 The reason for using MD as the chiral selector was simplicity and costs. MDs with three different dextrose equivalents (DE) were tested, and the obtained results showed a decreased resolution with increasing DE of MD. An MD with DE 4–7 dissolved in 100 mM phosphate buffer was chosen as the best chiral selector. The optimal concentration of MD was found to be 20% (w/v) and a buffer pH of 4.0 gave the lowest migration times and the best peak shapes. Besides the optimal conditions for CE separation and the detection, different EME parameters were investigated. As tolterodine was a hydrophobic tertiary amine, nitroaromatic solvents were tested as organic solvents. 2-Nitrophenyl octyl ether (NPOE) was found to be the best choice of organic solvent. The influence of other parameters, like voltage, extraction time, stirring rate, pH in acceptor phase and sample, was evaluated by an experimental design method. The best conditions were found to be as follows: 500 mM HCl as an acceptor phase; 10 mM HCl as the sample; 24 min extraction time; 54 V as potential difference; and 1200 rpm as stirring rate. The method was evaluated with respect to reproducibility (RSD), limit of quantification (LOQ), limit of detection (LOD), linearity (R2), dynamic linear range (DLR), and enrichment factor. Intra- and inter day RSD were less than 6.5%; LOQ and LOD for both enantiomers were 10 and 3 ng mL−1, respectively. R2 values were all above 0.9951 in the dynamic range 10–300 ng mL−1. The optimal extraction conditions were applied to determine S-tolterodine and R-tolterodine from water, urine and plasma samples, respectively. The extracts were injected into CE, and separation and detection of the enantiomers with the optimized MD-modified CE-UV method showed clean electropherograms with no visible interferences and acceptable recoveries.19
The lack of visible interferences described in the former work could be caused either by the selectivity of the extraction method (clean extracts) or by the selectivity of the detection method. UV is a relatively selective detection method, with high capability of detecting target analytes. However, selective detection methods like UV, MS and fluorescence only rarely provide any information regarding the interfering matrix components. To demonstrate the excellent clean-up properties of EME, the group of Kubáň, Boček and co-workers combined CE with the capacitively coupled contactless conductivity detection method (CE-C4D).21–23 In CE-C4D, all charged species are visualized conductometrically and is thus a more universal detection method compared to UV. This could be advantageous in combination with EME, where CE-C4D can be used for detection and monitoring of the extraction of both target analytes and interfering matrix components across the SLM, respectively.21 Three basic drugs (nortriptyline, haloperidol and loperamide) were chosen as target analytes: Na+, NH4+, K+, Ca+ and Mg2+ represented inorganic cations; the amino acids Crea, Lys and His were representatives for small biomolecules whereas the highly abundant human serum albumin (HSA) was selected as the large biomolecule. A CE-C4D method for separation and detection of the mentioned model analytes was developed and EME parameters were selected in accordance with earlier publications. Acetic acid was used as the acceptor phase and the sample to unite them with the background electrolyte (BGE) used in the CE separation. The EME-CE-C4D was used to examine the transfer of matrix components across the SLM, composed of different organic solvents (NPOE, ethyl nitrobenzene (ENB), 1-octanol and NPOE with addition of di(2-ethylhexyl)phosphate (DEHP), Fig. 4).
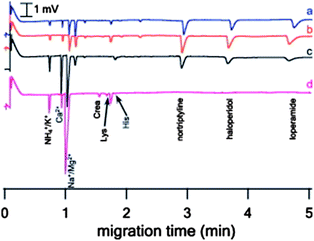 | ||
| Fig. 4 CE-C4D determination of EME pretreated standard solution using various SLMs. EME conditions: agitation: 800 rpm, extraction time: 10 min, extraction voltage: (a) NPOE (150 V), (b) ENB (10 V), (c) 1-octanol (5 V), and (d) NPOE–DEHP 85/15% v/v (25 V), acceptor solution: 100 mM acetic acid. Donor solution for EME was prepared in 1 M acetic acid. Concentration of basic drugs is 500 μg L−1. Reproduced with permission.21 | ||
Thus, a new tool for determination of SLM selectivity was developed. The method proved for the first time that macromolecules from plasma, like HSA, were effectively retained by all the SLMs included in the study, and that extraction of other matrix components across the SLM is strongly dependent on the nature and composition of the SLM. The main findings comprised a highly selective transfer of target analytes with pure NPOE as the SLM, whereas extraction of all the model molecules representing matrix components was suppressed. Addition of the anionic carrier molecule DEHP to NPOE increased the transfer of matrix components across the membrane while extraction of the target analytes was suppressed.
EME-CE-C4D has also been shown to be a useful method for determination of heavy metal cations from aqueous samples.22 Pb2+, Ni2+, Mn2+, Cd2+, Cu2+, Co2+, and Zn2+ were effectively extracted from aqueous samples with 1-octanol + 0.5% (v/v) DEHP as the SLM, 5 min extraction time, 750 rpm as stirring rate, and 75 V as the potential difference. The method was applied for determination of Zn2+ from tap water and powdered milk. The results from tap water were comparable to atomic absorbance spectroscopy (AAS) measurements, and good correlation was found between the concentrations in milk determined by the EME-CE-C4D method and the concentration given by the powdered milk manufacturer.
The same group used EME-CE-C4D for determination of amino acids from different body fluids.23 Because of the zwitterionic and hydrophilic nature of the amino acids, LLE with back extraction has traditionally been a difficult task to cope with.24 By usage of a tailored SLM in an EME set-up, 17 amino acids were effectively extracted from acidified human body fluids. EME in combination with CE-C4D proved that other possible interfering compounds were retained by the selective SLM. Endogenous levels of 12 of the amino acids were determined in human serum, plasma and whole blood. The proposed method was shown to be suitable for rapid and simple pre-treatment and measurement of elevated concentrations of selected amino acids, which could be possible biomarkers of severe inborn metabolic disorders. Determination of lithium from different human body fluids with EME-CE-C4D was also successfully performed by the same group.25
While the former method comprised an offline sample pre-treatment before injection into CE, another work performed by Kubáň et al. characterized an online coupling of the EME principle to CE-C4D.26 The SLM, consisting of 1-hexanol impregnated in a 100 μm thick polypropylene membrane, was clamped between two PTFE link chambers. One of the chambers was filled with the sample while the other functioned as the acceptor phase container. The device was coupled online with the CE instrument during the injection sequence. On sample injection, a platinum electrode coupled to a high voltage power supply was placed in the sample chamber, and the separation capillary touched the SLM in the acceptor chamber. Application of −10 kV for 30 seconds induced an electrokinetic injection across the SLM. After the injection, the separation capillary was removed from the pre-treatment device and placed into a vial containing the BGE buffer. The separation of the target analyte, perchlorate, was accomplished with a potential difference of −20 kV, and the target analytes were detected by C4D. Because of the SLM selectivity in the pre-treatment process, and because the universal detection method would disclose possible transfer of interferences, perchlorate was detected in a clean acceptor phase. The direct sample injection method across the SLM was tested on real samples. Perchlorate was determined from different spiked samples like human serum, human breast milk, red wine and tap water. The sensitivity of the online coupled SLM method was comparable to routinely used methods for perchlorate determination. However, the EME-CE-C4D was proposed as advantageous to existing methods with respect to the online combination, shorter run times, lower costs and no need for sample preparation.
Despite the speed, reduction in solvent consumption and disposal and the rapid method development offered by CE, this separation method is still characterized by relatively poor sensitivity and reproducibility compared with liquid chromatography (HPLC).27 Therefore, EME was coupled with HPLC-UV for the first time to obtain reproducible results when a theoretical model of the extraction process was developed.28 Five basic hydrophobic drugs (pethidine, nortriptyline, methadone, haloperidol and loperamide) were used as model analytes and extracted in a system with NPOE as the SLM. The extracts were injected into HPLC after a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dilution with hydrochloric acid in order to obtain a sufficient volume in the microinsert. The combination of EME with HPLC was successfully implemented as an alternative to EME-CE.
:1 dilution with hydrochloric acid in order to obtain a sufficient volume in the microinsert. The combination of EME with HPLC was successfully implemented as an alternative to EME-CE.
Several studies have demonstrated the excellent compatibility between EME and HPLC-UV, amongst other EME-HPLC-UV of target analytes from aqueous solutions,29 environmental samples,30 wastewater6,31–34 and from biological samples.5,7,8,33,35–37
To further push the detection limits and application range of EME, the extraction method has been combined with LC-MS in several studies. The first attempt to EME-LC-MS was carried out within the area of peptide analysis.38 The analytical performance of angiotensin I–III extractions from human plasma using optimized EME conditions was evaluated in combination with LC-MS, which is the preferred analytical technique for peptide analysis due to very selective and sensitive measurements.39 With an SLM consisting of 8% DEHP (w/w) in 1-octanol, 15 V potential difference across the SLM and with an extraction time of 10 minutes, the angiotensins spiked to human plasma were effectively extracted with recoveries between 25 and 43%. Due to the excellent sensitivity offered by LC-MS, detection limit of angiotensin II was determined to be 240 pg mL−1 with the proposed method. However, this LOD was higher than endogenous levels of angiotensin II. To enable the detection of endogenous levels of angiotensin II and other endogenous peptides of low abundance, the single quadrupole MS used in the former work was changed to a triple quadrupole MS for increased sensitivity.40 The reported LOD for angiotensin II was 60 pg mL−1, which is in the upper level of endogenous concentrations. With this approach, EME was used to detect low-abundance peptides in non-spiked human plasma for the first time.
The great possibility for identification of unknown peptides by MS/MS was exploited in another report.41 A complex mixture of 37 peptides obtained from tryptic digestion of cytochrome c and bovine serum albumin was extracted by EME and analysed on LC-MS/MS (Orbitrap). The reliable identification obtained by the MS/MS was used to study different extraction parameters of a mixture of peptides with a diverse range of physico-chemical properties. The results showed that large peptides were more difficult to extract compared to shorter peptides. However, both hydrophobic and hydrophilic peptides were effectively extracted by tailor-made SLMs.
Other examples of EME in combination with LC-MS include EME from undiluted human plasma,16,42 undiluted whole blood43 and from dried biological samples on water-soluble matrices44,45 and EME of quinolones from environmental water samples.46 The latter publications will be discussed in more details in the section “EME from diverse matrices”. EME with high enrichment factors from undiluted biological fluids in combination with the sensitivity offered by LC-MS gives very low LOQs. In one work, hydrophobic drugs were extracted exhaustively from 50 μL human plasma by increasing the number of hollow fibres from one to three, thus increasing the contact area between the sample and the SLM. The extracts were analysed by a single quadrupole MS, providing LOQs in the range 0.6–3.2 ng mL−1 for citalopram, loperamide, methadone, paroxetine, pethidine and sertraline, respectively.42 The LOQs were well below the normal therapeutic range of all the drugs, emphasizing the potential of EME-LC-MS for therapeutic drug monitoring. A follow-up work demonstrated the feasibility of EME-LC-MS, where EME of six basic drugs of abuse from undiluted whole blood and post mortem blood was reported.43 EME conditions were optimized on whole blood spiked with cathinone, methamphetamine, 3,4-methylenedioxy-amphetamine (MDA), 3,4-methylenedioxy-methamphetamine (MDMA), ketamine and 2,5-dimethoxy-4-iodoamphetamine (DOI). Within 5 min extraction time, an applied voltage of 15 V and 1-ethyl-2-nitrobenzene (ENB) as the SLM, the model analytes were extracted from 80 μL whole blood with recoveries in the range 10–30%. The optimized conditions were used to analyse real forensic whole blood samples taken from three forensic autopsy cases and on five forensic cases from living persons. The resulting chromatograms from the analysis on UPLC-MS/MS showed nicely the selective properties of EME-UPLC-MS/MS with MRM.
To further take advantage of the possible high enrichment factors by reduction of the acceptor phase volume, EME was combined with gas chromatography with flame ionization detection (GC-FID) in a recent publication.47 Two tricyclic antidepressants (imipramine and clomipramine) were used as model analytes. These basic, hydrophobic model analytes were effectively extracted within 20 min from 2.1 mL acidified sample solution (pH 4.0) through an SLM consisting of NPOE and into 6 μL of acceptor phase (10 mM HCl, pH 2.0). The model analytes were quantified by GC-FID, and because of the large sample:acceptor phase ratio (2100:6), enrichment factors between 270 and 280 were obtained. The performance of the method was demonstrated by simultaneous EME of the two model analytes from water, plasma and urine samples, with acceptable LODs, linearities and RSD values.
An alternative combination is two-phase EME followed by GC-MS determination. This approach was described for the first time by Davarani and co-workers.48 The extraction performance of four basic pharmaceutical compounds (imipramine, citalopram, desipramine and sertraline) in a two-phase system was investigated. The aqueous acceptor phase used in three-phase EME was exchanged with an organic solvent inside the lumen of the hollow fibre, and heptanol was found to be the best choice of organic solvent. Other parameters like effect of sample solution pH, effect of applied voltage, effect of stirring speed and extraction time were investigated, and the optimized method was thereafter compared with a three-phase EME procedure. With the proposed method, LOD was reduced from 0.8 ng mL−1 with three-phase EME-GC-FID47 to 0.1 ng mL−1 with two-phase EME-GC-MS.48 The authors also claimed that by using an organic solvent as the acceptor phase and thus making it GC-MS compatible, the applicability of EME is broaden. In addition, the presented work showed that ionic model substances were dissolved in the organic solvent under the presence of an applied electrical field, thus opening a new understanding of the basics of EME theory.
EME was also combined with GC-MS in another variant, where EME was followed by low-density solvent based ultrasound-assisted emulsification microextraction (LDS-USAEME) for determination of chlorophenols in water.49 In the first step, EME with 1-octanol as the SLM was performed on water samples. The aqueous acceptor phase was thereafter collected and transferred to another sample compartment, a plastic Pasteur pipette. In the second step, the chlorophenols were extracted into a low-density solvent (toluene) that was dispersed in the aqueous sample solution by the usage of ultrasound. After centrifugation, the organic solvent was collected and injected into a GC-MS system together with a derivatization reagent, MTBSTFA. The proposed method offered high enrichment factors (<2198), low LODs (0.005–0.0020 μg L−1) as well as acceptable linearity and repeatability. By usage of EME as the first clean-up step, some of the shortcomings of conventional USAEME regarding complex matrices were complied with.
The same group recently proposed electromembrane extraction-ion chromatography (EME-IC) as an alternative method for determination of biological organic anions.50 Ion chromatography in combination with EME has also been used for determination of inorganic anions in organic solvents like ethyl acetate,51 butyl acetate52 and liquefied petroleum gas.53
EME from diverse matrices
Since EME was published for the first time in 2006,1 several different matrices have been included in EME compatibility studies. The obtainable selectivity makes EME an attractive sample preparation method of complicated matrices dealt with in bioanalysis and environmental analysis, and the possibility of high enrichment factors and hence high degree of sensitivity is a great advantage within the same fields. However, the first publications focused on fundamental extraction parameters of EME from water samples spiked with basic drugs,4,11,28,54 acidic drugs12 and peptides.15,29 General factors such as shortened extraction times, pH dependence, magnitude of the applied voltage and tailored SLMs for increased selectivity were discussed. Because the innovative extraction method originally was developed for extraction from biological fluids, examples of EME from human plasma,1,7,13,14,17,19,25,37,38,55 urine,1,7,13,17,19,25,37,55,56 breast milk,13,55 saliva37,57 and amniotic fluids14 have been included in the experiments. The first systematic study describing EME from human plasma and whole blood without any sample pre-treatment like dilution or pH adjustment was published in 2009.58 It was earlier proved that the extraction performance in EME was relatively insensitive to the pH in the sample,1,12 and this phenomenon was exploited in extraction from pure biological fluids. Recoveries from EME from untreated whole blood spiked with six hydrophobic basic drugs ranged from 19 to 51%.58 The recoveries and the extraction kinetics were somewhat lower compared to untreated plasma and pure water samples because of higher viscosity and complexity of the whole blood samples compared to the others. However, the clean-up properties of EME from complex matrices were clearly demonstrated. This was also the focus of another publication, where trace amounts of the opioid antagonists nalmefene and naltrexone were extracted from untreated human plasma and urine with high degree of selectivity.5Another recent publication described the fast extraction kinetics of EME from untreated biological fluids in a stagnant system.16 Four basic drugs were quantified in plasma within only one minute of extraction time, with a common battery as the power supply (9 V) and without any convection of the system. This extremely simple set-up was demonstrated on untreated whole blood and urine as the sample matrix.
Other studies that cover EME from undiluted biological samples include exhaustive extraction of citalopram, loperamide, methadone, paroxetine, pethidine, and sertraline42 and EME of trimipramine enantiomers.18
A biological matrix that is even more complicated than whole blood in a sample preparation perspective is post-mortem blood.59 Due to putrefaction or trauma of the body during the dead process, isolation of substances from post-mortem matrices is in general more difficult than from other clinical specimens. EME has recently shown to be a valuable tool in that respect.43 As mentioned in an earlier section, six basic drugs of abuse were extracted from whole blood and post-mortem blood, and the extracts analysed by LC-MS/MS were very clean despite the dirty and complex matrix.
A newly published invention is about EME in combination with dried matrix spots (DMS), described for dried blood spots44 and dried oral spots,45 respectively. In both cases, the sample matrix consisted of biological fluids dried on a water-soluble, polymeric material, thereafter resolved in an acidified aqueous solution. Thus, the resulting matrix contained all the components in the original biological fluid and solubilized polymer material (alginate or chitosan). The analytes of interest were selectively extracted from this matrix by usage of optimized EME. The resulting chromatograms obtained after LC-MS of the extracts did not contain any co-extracted interferences, and there was no indication of ion suppression in any of the two published cases. With the new material suggested for DMS in combination with EME, the recoveries were found to be higher in comparison to the recoveries obtained with the manufacturer's procedure on commercial cards.44
In order to increase the compatibility between EME and commercially available DMS cards, a new study focused on EME from samples with high content of organic solvent.60 The research indicated that EME with NPOE as the supported liquid membrane was compatible with samples containing up to 50% of ethanol or methanol, or up to 75% dimethyl sulfoxide (DMSO), respectively. With EME from matrices with acetonitrile, a stable system was not obtained, as the matrix partly dissolved the SLM during extraction. The findings were used to test EME from dried blood spot eluates originally containing 80% methanol.
Apart from excellent extraction properties from biological matrices, EME has also shown to be usable for sample preparation from wastewater due to its selectivity and sensitivity properties. Different substances, like acidic drugs,6,33,61,62 basic drugs,32,61 haloacetic acids and aromatic acetic acids31 and fluoroquinolones,34 have been extracted from wastewater with high degree of clean-up and enrichment. Other applications from environmental samples include EME of chlorophenols from sea water30 and drain water,49 phenoxy acid herbicides from river water,20 quinolones from underground water, sea water and river water,46 lipophilic anions from river water63 and trace determination of perchlorate from snow and drinking water.64
Improvements of EME performance
Lots of effort have been made in the last few years to bring the knowledge of EME from a limited, fundamental level to an expanded theoretical understanding and to new application areas. While the former sections mostly described new applications, this section will focus on the recent ways to improve the EME performance.Simultaneous extraction of anions and cations
Extraction of anionic and cationic substances in one single sample preparation step is a challenging task. This challenge has been addressed by EME in several publications,56,61,65 and one technical solution to this is illustrated in Fig. 5. The set-up utilized two separate hollow fibres inserted into the sample. EME was performed with neutral pH in the sample, where both the acidic drug diclofenac and the basic drug nalmefene were ionized. Diclofenac was extracted into the hollow fibre containing the positive electrode. In this hollow fibre, 1-octanol was used as the SLM, and the acceptor phase was 50 mM NaOH. Nalmefene was extracted into the other hollow fibre which contained the negative electrode. Here, 2-nitrophenyl octyl ether with 5% di-(2-ethylhexyl) phosphate was used as SLM, and the acceptor phase was 50 mM HCl. Thus, both acidic and basic substances were extracted simultaneously, and were isolated into two separate acceptor phases. This idea may open up a very interesting direction for future EME.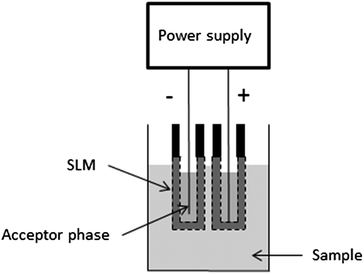 | ||
| Fig. 5 Schematic illustration of set-up used for simultaneous EME of anions and cations. | ||
New membrane developments
One of the main focus areas in the development of EME has been the supported liquid membrane and how it works together with the applied voltage. Tailored membranes have been shown to give very selective extractions.11,41,66 However, there is still a lack of theoretical knowledge about the role of the SLM in the extraction process. To meet this demand, a recent publication described a large screening of differently composed SLMs, both pure organic solvents and organic solvents with additives.67 The extraction efficiency of the SLMs was tested on eight model peptides, and the work identified mono- or disubstituted phosphate groups as effective carrier molecules in combination with either primary alcohols and ketones.A new way of thinking about the material supporting the organic solvent was described by Hasheminasab et al.62 In this innovative work, the commonly used hollow fibre made of polypropylene was replaced by a carbon nanotube (CNT) reinforced hollow fibre. The new sorbent interface was used to extract two acidic nonsteroid anti-inflammatory drugs (NSAIDs) (ibuprofen and naproxen) from different biological and environmental matrices. Because of the large surface area and high adsorption capacity offered by the CNTs, resulting in an increase of the overall analyte partition coefficient, excellent preconcentration factors (PF = 180–188) and recoveries (90–94%) were obtained compared to a conventional EME method. Similar results were shown in a parallel study performed by the same group, where buprenorphine was extracted from urine samples by CNT-EME.68
One challenge to the way of making traditional SLMs is the mechanical robustness of the SLM.69 Unless the water-solubility of the organic solvent is extremely low, some of the SLM will leak into the sample under agitation and application of electrical field. This was addressed by the group of See and Hauser, which demonstrated EME of lipophilic anions through a so-called carrier-mediated polymer inclusion membrane (PIM) for the first time.63 PIMs are self-supporting membranes, where a base polymer, a plasticizer and an eventual functional carrier are incorporated into a homogenous membrane. The optimized PIM consisted of 60% cellulose triacetate, 20% 2-nitrophenyl octylether (NPOE) and 20% Aliquat 336. By application of 700 V across the PIM, the three model anions propanesulfonate, octanesulfonate, and decanesulfonate were effectively extracted from spiked river water samples.
The last contribution to the field of improved EME membranes described an electrically driven facilitated transport of Cs+ across copper ferrocyanide channels in track etched membrane.70 An ion selective inorganic exchanger, copper ferrocyanide, was loaded into the pores of a polycarbonate track etched membrane (PTEM). When low potential differences were applied, Cs+ cations were selectively transported across the membrane.
Developments in the electrical field application
The driving force in EME was originally a constant voltage applied across the supported liquid membrane.1 Several groups have studied the behaviour of the applied voltage and suggested different variants of the application. The group of Boček et al. supposed an improvement of the extraction performance by usage of a stabilized constant dc electric current instead of constant voltage.71 This invention reduced the RSD values from 3.6–17.8% at constant voltage to 2.8–8.9% for EME at constant current.Another variant comprised a pulsed voltage in EME instead of constant voltage.72 The new approach, termed pulsed electromembrane extraction (PEME), was claimed to be a more stable extraction system compared to conventional EME. A similar study was performed by the same group, which did a one-way and two-way pulsed EME for trace analysis of amino acids in food and biological samples.73
EME in combination with other sample preparation techniques
EME has been proposed as an excellent clean-up step in combination with conventional sample preparation method. The group of Lee et al. combined EME with a second step of low-density solvent based ultrasound-assisted emulsification microextraction (EME-LDS-USAEME).49 With this two-step method, high enrichment factors of up to 2198, low limits of detection (>0.005 μg L−1) and good linearity were achieved.In parallel to this, another study focused on the combination of EME with dispersive liquid–liquid microextraction (DLLME).74 The sensitive method was used for determination of tricyclic antidepressants (TCAs) present in low concentrations in human plasma and urine.
An interesting procedure was suggested by Rezazadeh et al.75 Inside the lumen of the hollow fibre, they used a carbonaceous pencil lead as the cathode, which simultaneously acted as a solid phase microextraction sorbent (Fig. 6). Subsequent to the extraction, the model analytes amitriptyline and doxepin were thermally desorbed into a GC injection port. This novel approach was termed electromembrane surrounded solid phase microextraction (EME-SPME), and was claimed to be a very efficient extraction method from complicated matrices.
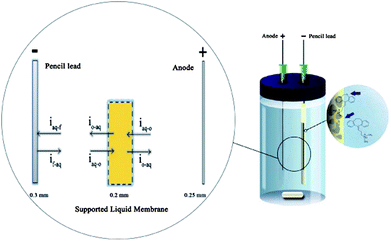 | ||
| Fig. 6 Equipment used for the EME-SPME method and mechanism of transport across liquid–liquid–solid boundaries. The flux of the analytes is represented by “i”; and o, aq, and f represent the organic, the aqueous and the carbonaceous fiber, respectively. Reproduced with permission.75 | ||
Future perspectives and conclusions
The scientific reports reviewed in this paper have demonstrated several interesting aspects related to electromembrane extraction (EME). EME can be accomplished with very simple and low-cost equipment, and the consumption of hazardous organic solvents can be reduced to only a few μL per sample. Extractions can normally be finished within 5 minutes, and give excellent sample clean-up even from complicated biological and environmental samples.Although EME is promising in several aspects, development of commercially available equipment is mandatory for the future. Currently, all EME experiments have been accomplished with home-built equipment, and this limits the implementation of EME into more research laboratories. However, work is in progress to commercialize the technology, and hopefully this will be finalized in a short time. In addition, more systematic knowledge should be developed related to tailoring different supported liquid membranes to different specific applications. The knowledge on supported liquid membranes is currently limited, and most existing work has the character of “looking for a needle in the haystack”. However, some recent and currently unpublished results in our laboratory have indicated some very interesting and strong selectivity phenomena, which have never been observed or reported in the literature. These phenomena are currently under systematic investigation, and are probably related to the fact that the behaviour of the organic solvent used as the SLM is highly influenced by the electrical field.
With commercial equipment for EME and with highly selective and tailored SLMs in the future, EME will hopefully be a valuable tool for the analytical laboratory. Competing with LLE and SPE in existing standard applications will be difficult, but there should be space for EME in new and more specialized applications, or in very complicated applications where the success of LLE and SPE is currently limited.
Notes and references
- S. Pedersen-Bjergaard and K. E. Rasmussen, J. Chromatogr., A, 2006, 1109, 183–190 CrossRef CAS
.
- S. Pedersen-Bjergaard and K. E. Rasmussen, Anal. Chem., 1999, 71, 2650–2656 CrossRef CAS
- M. Ghambarian, Y. Yamini and A. Esrafili, Microchim. Acta, 2012, 177, 271–294 CrossRef CAS
- A. Gjelstad, T. M. Andersen, K. E. Rasmussen and S. Pedersen-Bjergaard, J. Chromatogr., A, 2007, 1157, 38–45 CrossRef CAS
- M. Rezazadeh, Y. Yamini and S. Seidi, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2011, 879, 1143–1148 CrossRef CAS
- M. R. Payán, M. Á. B. López, R. F. Torres, M. V. Navarro and M. C. Mochón, Talanta, 2011, 85, 394–399 CrossRef
- M. Eskandari, Y. Yamini, L. Fotouhi and S. Seidi, J. Pharm. Biomed. Sci., 2011, 54, 1173–1179 CrossRef CAS
- L. Fotouhi, Y. Yamini, S. Molaei and S. Seidi, J. Chromatogr., A, 2011, 1218, 8581–8586 CrossRef CAS
- A. Gjelstad, H. Jensen, K. E. Rasmussen and S. Pedersen-Bjergaard, Anal. Chim. Acta, 2012, 742, 10–16 CrossRef CAS
- K. F. Seip, H. Jensen, M. H. Sønsteby, A. Gjelstad and S. Pedersen-Bjergaard, Electrophoresis, 2013, 34, 792–799 CrossRef CAS
- A. Gjelstad, K. E. Rasmussen and S. Pedersen-Bjergaard, J. Chromatogr., A, 2006, 1124, 29–34 CrossRef CAS
- M. Balchen, A. Gjelstad, K. E. Rasmussen and S. Pedersen-Bjergaard, J. Chromatogr., A, 2007, 1152, 220–225 CrossRef CAS
- I. J. O. Kjelsen, A. Gjelstad, K. E. Rasmussen and S. Pedersen-Bjergaard, J. Chromatogr., A, 2008, 1180, 1–9 CrossRef CAS
- C. Basheer, S. H. Tan and H. K. Lee, J. Chromatogr., A, 2008, 1213, 14–18 CrossRef CAS
- M. Balchen, H. Jensen, L. Reubsaet and S. Pedersen-Bjergaard, J. Sep. Sci., 2010, 33, 1665–1672 CrossRef CAS
- L. E. E. Eibak, A. Gjelstad, K. E. Rasmussen and S. Pedersen-Bjergaard, J. Chromatogr., A, 2010, 1217, 5050–5056 CrossRef CAS
- S. Nojavan and A. R. Fakhari, J. Sep. Sci., 2010, 33, 3231–3238 CrossRef CAS
- A. R. Fakhari, H. Tabani, S. Nojavan and H. Abedi, Electrophoresis, 2012, 33, 506–515 CrossRef CAS
- A. R. Fakhari, H. Tabani, H. Behdad, S. Nojavan and M. Taghizadeh, Microchem. J., 2013, 106, 186–193 CrossRef CAS
- H. Tabani, A. R. Fakhari and E. Zand, Anal. Methods, 2013, 5, 1548–1555 RSC
- P. Kubáň and P. Boček, J. Chromatogr., A, 2012, 1267, 96–101 CrossRef
- P. Kubáň, L. Strieglerova, P. Gebauer and P. Boček, Electrophoresis, 2011, 32, 1025–1032 CrossRef
- L. Strieglerová, P. Kubáň and P. Boček, J. Chromatogr., A, 2011, 1218, 6248–6255 CrossRef
- Y. K. Park, K. Choi, A. Y. B. H. Ahmed, Z. A. Alothman and D. S. Chung, J. Chromatogr., A, 2010, 1217, 3357–3361 CrossRef CAS
- L. Strieglerová, P. Kubáň and P. Boček, Electrophoresis, 2011, 32, 1182–1189 CrossRef
- P. Kubáň, I. K. Kiplagat and P. Boček, Electrophoresis, 2012, 33, 2695–2702 CrossRef
- L. Suntornsuk, Anal. Bioanal. Chem., 2010, 398, 29–52 CrossRef CAS
- A. Gjelstad, K. E. Rasmussen and S. Pedersen-Bjergaard, J. Chromatogr., A, 2007, 1174, 104–111 CrossRef CAS
- M. Balchen, L. Reubsaet and S. Pedersen-Bjergaard, J. Chromatogr., A, 2008, 1194, 143–149 CrossRef CAS
- J. Lee, F. Khalilian, H. Bagheri and H. K. Lee, J. Chromatogr., A, 2009, 1216, 7687–7693 CrossRef CAS
- K. Alhooshani, C. Basheer, J. Kaur, A. Gjelstad, K. E. Rasmussen, S. Pedersen-Bjergaard and H. K. Lee, Talanta, 2011, 86, 109–113 CrossRef CAS
- M. Rezazadeh, Y. Yamini and S. Seidi, J. Sep. Sci., 2012, 35, 571–579 CrossRef CAS
- S. S. H. Davarani, A. Pourahadi, S. Nojavan, M. H. Banitaba and M. Nasiri-Aghdam, Anal. Chim. Acta, 2012, 722, 55–62 CrossRef CAS
- M. Ramos-Payán, M. Villar-Navarro, R. Fernández-Torres, M. Callejón-Mochón and M. Bello-López, Anal. Bioanal. Chem., 2013, 405, 2575–2584 CrossRef
- S. Seidi, Y. Yamini, A. Heydari, M. Moradi, A. Esrafili and M. Rezazadeh, Anal. Chim. Acta, 2011, 701, 181–188 CrossRef CAS
- S. Seidi, Y. Yamini, T. Baheri and R. Feizbakhsh, J. Chromatogr., A, 2011, 1218, 3958–3965 CrossRef CAS
- S. Seidi, Y. Yamini, A. Saleh and M. Moradi, J. Sep. Sci., 2011, 34, 585–593 CrossRef CAS
- M. Balchen, T. G. Halvorsen, L. Reubsaet and S. Pedersen-Bjergaard, J. Chromatogr., A, 2009, 1216, 6900–6905 CrossRef CAS
- I. van den Broek, R. W. Sparidans, J. H. M. Schellens and J. H. Beijnen, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2008, 872, 1–22 CrossRef CAS
- M. Balchen, H. Lund, L. Reubsaet and S. Pedersen-Bjergaard, Anal. Chim. Acta, 2012, 716, 16–23 CrossRef CAS
- M. Balchen, A. G. Hatterud, L. Reubsaet and S. Pedersen-Bjergaard, J. Sep. Sci., 2011, 34, 186–195 CrossRef CAS
- L. E. E. Eibak, A. Gjelstad, K. E. Rasmussen and S. Pedersen-Bjergaard, J. Pharm. Biomed. Sci., 2012, 57, 33–38 CrossRef
- R. E. G. Jamt, A. Gjelstad, L. E. E. Eibak, E. L. Øiestad, A. S. Christophersen, K. E. Rasmussen and S. Pedersen-Bjergaard, J. Chromatogr., A, 2012, 1232, 27–36 CrossRef CAS
- L. E. E. Eibak, A. B. Hegge, K. E. Rasmussen, S. Pedersen-Bjergaard and A. Gjelstad, Anal. Chem., 2012, 84, 8783–8789 CrossRef CAS
- C. P. Lødøen, L. E. Eng Eibak, K. E. Rasmussen, S. Pedersen-Bjergaard, T. Andersen and A. Gjelstad, Bioanalysis, 2013, 5, 317–325 CrossRef
- M. H. Wang and S. P. Wang, J. Sep. Sci., 2012, 35, 702–706 CrossRef CAS
- S.
S. H. Davarani, A. M. Najarian, S. Nojavan and M. A. Tabatabaei, Anal. Chim. Acta, 2012, 725, 51–56 CrossRef CAS
- S. S. H. Davarani, A. Morteza-Najarian, S. Nojavan, A. Pourahadi and M. B. Abbassi, J. Sep. Sci., 2013, 36, 736–743 CrossRef CAS
- L. Guo and H. K. Lee, J. Chromatogr., A, 2012, 1243, 14–22 CrossRef CAS
- T. Y. Tan, C. Basheer, K. P. Ng and H. K. Lee, Anal. Chim. Acta, 2012, 739, 31–36 CrossRef CAS
- Z. Z. Hu, H. D. Chen, C. Y. Yao and Y. Zhu, J. Chromatogr. Sci., 2011, 49, 617–621 CAS
- Z. Z. Hu, L. Wang, C. Y. Yao, Y. Zhu and P. M. Zhang, Chin. J. Anal. Chem., 2011, 39, 1261–1265 CAS
- M. L. Li, J. M. Yang, H. F. Li and J. M. Lin, J. Sep. Sci., 2012, 35, 1365–1371 CrossRef CAS
- T. M. Middelthon-Bruer, A. Gjelstad, K. E. Rasmussen and S. Pedersen-Bjergaard, J. Sep. Sci., 2008, 31, 753–759 CrossRef CAS
- N. C. Dominguez, A. Gjelstad, A. M. Nadal, H. Jensen, N. J. Petersen, S. H. Hansen, K. E. Rasmussen and S. Pedersen-Bjergaard, J. Chromatogr., A, 2012, 1248, 48–54 CrossRef CAS
- S. Seidi, Y. Yamini, M. Rezazadeh and A. Esrafili, J. Chromatogr., A, 2012, 1243, 6–13 CrossRef CAS
- S. Seidi, Y. Yamini and M. Rezazadeh, J. Pharm. Biomed. Sci., 2011, 56, 859–866 CrossRef CAS
- A. Gjelstad, K. E. Rasmussen and S. Pedersen-Bjergaard, Anal. Bioanal. Chem., 2009, 393, 921–928 CrossRef CAS
- O. Drummer, Anal. Bioanal. Chem., 2007, 388, 1495–1503 CrossRef CAS
- K. F. Seip, A. Gjelstad and S. Pedersen-Bjergaard, J. Chromatogr., A, 2013 Search PubMed
- C. Basheer, J. Lee, S. Pedersen-Bjergaard, K. E. Rasmussen and H. K. Lee, J. Chromatogr., A, 2010, 1217, 6661–6667 CrossRef CAS
- K. S. Hasheminasab, A. R. Fakhari, A. Shahsavani and H. Ahmar, J. Chromatogr., A, 2013, 1285, 1–6 CrossRef CAS
- H. H. See and P. C. Hauser, Anal. Chem., 2011, 83, 7507–7513 CrossRef CAS
- I. K. Kiplagat, T. K. O. Doan, P. Kubáň and P. Boček, Electrophoresis, 2011, 32, 3008–3015 CrossRef CAS
- H. Tabani, A. R. Fakhari and A. Shahsavani, Electrophoresis, 2013, 34, 269–276 CrossRef CAS
- P. Kubáň, A. Šlampová and P. Boček, Electrophoresis, 2010, 31, 768–785 CrossRef
- K. F. Seip, J. Stigsson, A. Gjelstad, M. Balchen and S. Pedersen-Bjergaard, J. Sep. Sci., 2011, 34, 3410–3417 CrossRef CAS
- K. S. Hasheminasab and A. R. Fakhari, Anal. Chim. Acta, 2013, 767, 75–80 CrossRef CAS
- A. K. Pabby and A. M. Sastre, J. Membr. Sci., 2013, 430, 263–303 CrossRef CAS
- S. Chaudhury, C. Agarwal, A. K. Pandey, A. Goswami and P. U. Sastry, J. Membr. Sci., 2013, 434, 93–98 CrossRef CAS
- A. Slampova, P. Kuban and P. Bocek, J. Chromatogr., A, 2012, 1234, 32–37 CrossRef CAS
- M. Rezazadeh, Y. Yamini, S. Seidi and A. Esrafili, J. Chromatogr., A, 2012, 1262, 214–218 CrossRef CAS
- M. Rezazadeh, Y. Yamini, S. Seidi and A. Esrafili, Anal. Chim. Acta, 2013, 773, 52–59 CrossRef CAS
- S. Seidi, Y. Yamini and M. Rezazadeh, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2013, 913–914, 138–146 CrossRef CAS
- M. Rezazadeh, Y. Yamini, S. Seidi and B. Ebrahimpour, J. Chromatogr., A, 2013, 1280, 16–22 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2013 |