Detection of the organophosphorus nerve agent VX and its hydrolysis products in white mustard plants grown in contaminated soil
Matthew R.
Gravett
*a,
Farrha B.
Hopkins
a,
Marcus J.
Main
a,
Adam J.
Self
a,
Christopher M.
Timperley
a,
Andrew J.
Webb
a and
Matthew J.
Baker
*ab
aDetection Department, Defence Science and Technology Laboratory (Dstl), Porton Down, Salisbury, Wiltshire SP4 0JQ, UK. E-mail: mrgravett@dstl.gov.uk
bCentre for Materials Science, J B Firth Building, Division of Chemistry, University of Central Lancashire, Preston, Lancashire PR1 2HE, UK. E-mail: mjbaker@uclan.ac.uk
First published on 1st October 2012
Abstract
The Chemical Weapons Convention (CWC) aims to prohibit the development, production, acquisition, stockpiling, retention, transfer or use of chemical weapons by States Parties. Verification of compliance or investigations into allegations of use requires accurate detection of chemical warfare agents (CWAs) and their breakdown products. Detection of CWAs such as organophosphorus nerve agents in the environment relies mainly upon the analysis of soil. Here we present a novel method for the detection of the nerve agent VX and its hydrolysis products through analysis using a combination of gas chromatography-mass spectrometry (GC-MS) and liquid chromatography-mass spectrometry (LC-MS) of ethanol extracts of contaminated vegetation (white mustard, Sinapis alba), which localised the compounds of interest, and in this study retained them in an extractable form longer than the soil.
Introduction
Chemical warfare agents (CWAs) have been condemned since their first use during World War I.1 Most recently, they were used by Iraq during the First Gulf War2 and by the Aum Shinrikyo cult in Matsumoto in 1994 and on the Tokyo underground in 1995.3 The international community through the Organisation for the Prohibition of Chemical Weapons (OPCW) in The Hague, which implements the CWC, aims to eliminate chemical weapons.4 Analysis of CWAs and their degradation products is essential for verification of compliance to the CWC and for confirming CWA use. Gas chromatography (GC) and liquid chromatography (LC), combined with mass spectrometry (MS), are the main techniques used to detect and identify these chemicals.5 Using both GC and LC-MS methodologies extends the range of compounds, from a single sample, that are detectable due to differences in the volatilities of the analytes and ionisation mechanisms for each technique. The UK National Security Strategy highlights international terrorism affecting the UK or its interests, including a chemical attack by terrorists, as a Tier One risk6 and the US views countering the proliferation of chemical weapons technology as a priority for defence in the 21st century.7Nerve agents are organophosphorus chemicals that inhibit the enzyme acetylcholinesterase, disturbing transmission of nerve impulses.3 Chief among them are O-ethyl S-2-diisopropylaminoethyl methylphosphonothiolate (VX)8 and O-isopropyl methylphosphonofluoridate (sarin). These compounds react with moisture to yield the alkyl methylphosphonic acids EMPA or iPMPA9 and more slowly, methylphosphonic acid, MPA10 (Fig. 1). Such products indicate the prior presence of the nerve agents. Sarin has greater volatility and reactivity to water than VX, and persists for a shorter time in the environment.11,12 The hydrolysis products EMPA, iPMPA and MPA are water-soluble and are retained by soil according to its composition, which varies widely.13 Vegetation may absorb nerve agents and their degradation products, acting as a time capsule whose interrogation can reveal nerve agent use. The literature lacks proof for the uptake of CWAs by vegetation grown in contaminated soil, although the nerve agent soman is absorbed by wheat in hydroponic culture.14
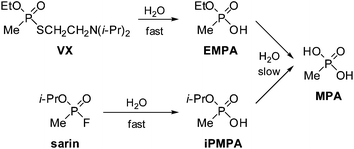 | ||
| Fig. 1 The nerve agents VX and sarin react with water in the environment to provide ethyl methylphosphonic acid (EMPA) or isopropyl methylphosphonic acid (iPMPA). These acids react slowly with water with loss of either ethanol or isopropanol to provide methylphosphonic acid (MPA). | ||
For the analysis of environmental samples associated with alleged use, where analyte concentrations may be in the low parts per billion range, derivatisation may be used to enhance the selectivity or sensitivity of detection of the phosphonic acid degradation products (e.g. EMPA, iPMPA and MPA). Conversion to trimethylsilyl derivatives is the approved procedure for OPCW on-site analysis during inspections. For off-site analysis, some OPCW designated laboratories prefer to prepare tert-butyldimethylsilyl (TBDMS) derivatives. These have the advantage of greater resistance to hydrolysis and stability on storage. Both the silylating agents and derivatives produced are sensitive to moisture. Consequently current recommended sample preparation procedures require concentration of aqueous extracts or solutions of degradation products to dryness prior to derivatisation. Residues are derivatised in an organic solvent usually with heating, particularly for the TBDMS derivatives, and analysed by GC using generic or selective detectors, or preferably by GC-MS.
The detection of CWAs by designated national laboratories is a requirement of the CWC and supports national security strategies. The Defence Science and Technology Laboratory (Dstl) at Porton Down has housed the UK Designated Laboratory since the CWC entered into force in 1997. An ability to unequivocally identify CWAs, their precursors and degradation products, in various environmental matrices, at concentrations ranging from neat material to parts per billion, is an essential capability.10 We now present a novel detection methodology involving ethanol extraction of white mustard, Sinapis alba, grown in soil contaminated with VX and iPMPA (to simulate sarin use) and analysis of the extracts by GC-MS after TBDMS derivatisation of the phosphonic acid degradation products. Currently, CWA use is determined through analysis of soil and debris suspected of contamination. Successful detection depends upon the sampling regime and efficient extraction and sample preparation. A system that localises and stores CWAs, that is amenable to extraction, should greatly enhance the ability to attribute CWA use.
Materials and methods
Spiking chemicals
O-Ethyl S-2-diisopropylaminoethyl methylphosphonothiolate (VX) and O-isopropyl methylphosphonic acid (iPMPA) of 98% purity by NMR spectroscopy (1H, 13C and 31P nuclei) were prepared at Dstl Porton Down.8,9 Methylphosphonic acid of 98% purity was from Sigma-Aldrich Ltd. (Dorset, UK).Plant cultivation and harvest
Sinapis alba is pollution-tolerant plant15 that originates from the Mediterranean floral region. Nowadays it grows wild cosmopolitically and in cultivation, reaching dimensions of 60 cm × 30 cm in sand, loam and heavy soils of acidic, basic and neutral pH.16 It is grown for its edible leaves and seeds, and as a green manure due to its rapid growth and coverage.A standard seed tray (Grow It, 20 cell insert, Gardman, Lincs., UK) was levelly filled to the top with soil (Levington Seed & Cutting Compost, The Scotts Miracle-Gro Company, Surrey, UK). A single divot was created in each compartment and one seed was placed into each divot. The seed was covered with soil and contaminated with 1 ml of a 250 μg ml−1 aqueous solution of VX or iPMPA. The soil was watered with local borehole water (10 ml) immediately and then at 24 h intervals. The trays were placed under a lighting system (EvoLux Bright-Wing Mother Clone Lights, Growell, UK) that provided 38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 lumens covering an area of 240 cm × 240 cm and on a timing system to provide 10 h of light every 24 h. Four plants were harvested at each of four time intervals (5, 9, 16 and 28 days). The plants were removed from the soil, and both plants and soil were immediately frozen and stored at −40 °C in a freezer. Control samples of soil, and soil with seeds, were cultivated similarly without contamination by CWA solutions.
400 lumens covering an area of 240 cm × 240 cm and on a timing system to provide 10 h of light every 24 h. Four plants were harvested at each of four time intervals (5, 9, 16 and 28 days). The plants were removed from the soil, and both plants and soil were immediately frozen and stored at −40 °C in a freezer. Control samples of soil, and soil with seeds, were cultivated similarly without contamination by CWA solutions.
Plant sample preparation
The plant samples were defrosted and pulverised using a pestle and mortar and extracted with ethanol (4 ml, 96% v/v, BDH AnalaR, VWR International Ltd., West Sussex, UK) and the pestle and mortar rinsed with ethanol (3 × 2 ml). The combined extracts were filtered using a 0.45 μm nylon filter (Whatman, Kent, UK) and concentrated under nitrogen gas to a volume of 1 ml. A sample (100 μl) of this was analysed by LC-MS (Q-ToF, Agilent) while another aliquot (100 μl) was dried at 35 °C for 2 h using a SpeedVac (Thermo Scientific Ltd., UK). Derivatisation was performed using N-tert-butyldimethylsilyl-N-methyltrifluoroacetamide (MTBSTFA) and 1% tert-butyldimethylchlorosilane (TBDMSCI, 50 μl; Sigma-Aldrich Ltd.), pyridine (5 μl; Pierce, silylation grade) and acetonitrile (45 μl; Thermo Scientific Ltd., silylation grade). The solution was heated at 100 °C for 30 min (Regis Technologies Inc., IL, USA) and analysed by GC-MS (6890-5973, Agilent, UK).Soil sample preparation
The soil samples were defrosted, extracted with ethanol (40 ml), filtered using a 0.45 μm nylon filter, concentrated to 2 ml and subsequently filtered using a 0.45 μm nylon filter. An aliquot (100 μl) was analysed by LC-MS, while 1 ml was taken to dryness and derivatised for GC-MS analysis as before. Separate soil samples were spiked with aqueous solutions of EMPA, MPA and iPMPA at concentrations of 10 μg g−1 and the ethanol extraction and analysis procedure already described followed. Extraction with 1 M aqueous NaOH (1 ml) and ultra-high purity water (20 ml) was also performed and the extracted sample processed as before.Instrumentation and analysis
GC-MS was performed on an Agilent Technologies 6890GC-5973 mass spectrometer (detection limit 1 pg μl−1 of octafluoronaphthalene) equipped with an autosampler. The GC was performed in splitless mode with the injection port kept at 250 °C and 45.6 ml min−1 purge of helium. The initial temperature of the GC oven (90 °C) was held for 0.7 min and ramped up 10 °C min−1, to 300 °C, where it was held for 2 min. An Agilent Technologies J&W DB-5MS (25 m in length, diameter of 0.2 mm, and a film thickness of 0.33 μm) was used in constant flow mode with a flow of 1.0 ml min−1 helium. The total run time was 23.70 min and the injection volume was 1 μl. External calibration standards of derivatised iPMPA (10 μg ml−1), EMPA (10 μg ml−1) and MPA (10 μg ml−1) were used.LC-MS was performed on an Agilent Technologies 6530 Q-ToF mass spectrometer (detection limit 1 pg on column of reserpine) equipped with an Agilent Technologies Infinity 1290 HPLC pump and autosampler. The mass spectrometer was operated in positive-ion Agilent Jet Stream Electrospray Ionisation (AJS-ESI) mode. The LC column was a Zorbax Eclipse Plus C18 Rapid Resolution HD (Agilent Technologies) of length 50 mm, internal diameter 2.1 mm, and particle size 1.8 μm. A 1290 Infinity inline filter (0.3 μm) (Agilent Technologies) was fitted to the column inlet. Mobile phases were: A (0.1% formic acid in water) and B (0.1% formic acid in acetonitrile). The elution gradient was 5% B (0.0–3.5 min) to 50% B (3.5–4.0 min) to 100% B (4.0–5.0 min) at a flow rate of 1 ml min−1. AJS-ESI-MS conditions were: drying gas temperature (nitrogen) 300 °C at 4 l min−1, nebuliser PSIG, sheath gas temperature (nitrogen) 300 °C at 12 l min−1, capillary voltage 4000 V, nozzle voltage 1000 V, MS ToF fragmentor 175 V, skimmer 65 V, octopole 1 RF Vpp 750 V, scan range m/z 50–1700, and scan time 0.33 s. An injection volume of 10 μl and an external calibration standard of 1 μg ml−1 aqueous VX was used.
Data analysis
Initial data analysis was performed using MassHunter (Agilent Technologies). Data was exported to Microsoft Excel for quantification and graphical representation. Quantification was based upon extracted ions; m/z 268.14946 for VX, m/z 153 for iPMPA and EMPA, and m/z 267 for MPA. Concentration results for each time point represent the average of four plant analyses.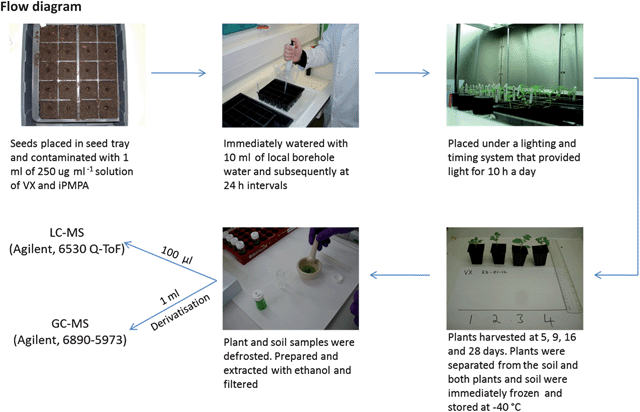
Results and discussion
The VX concentration in the plants increased up to day 9 and then fell by day 28 (Fig. 2). A similar profile was observed for the degradation products EMPA and MPA. The concentration of VX in the soil remained constant until day 9 and then plummeted by day 16, falling further by day 28. This is consistent with other studies of VX on soil17 where VX hydrolysis, once started, occurs autocatalytically.18 The VX concentration in the plant increased while VX was available in the soil for uptake, but decreased as the VX concentration in the soil declined. The degradation products of VX are difficult to detect in soils with a high organic content, such as the Levington compost used, due to absorption and retention of VX. This was confirmed by a soil spiking study where EMPA and MPA were added to the soil and then extracted by two techniques: (a) with ethanol, or (b) by basification of the sample, extraction with ultra-high purity water, and derivatisation as before. In both cases, it was not possible to detect EMPA and MPA. However, using Sinapis alba, we were able to overcome this problem, with a simple ethanol extraction, and detect both EMPA and MPA at day 28. This methodology would be able to provide evidence of use of VX that would otherwise be unavailable using standard procedures. | ||
| Fig. 2 Concentration of VX and its degradation products in Sinapis alba and soil. (A) VX concentration profile in Sinapis alba, (B) phosphonic acid concentration in Sinapis alba, and (C) VX concentration in soil. Results for each time point represent an average of analyses of four plants. | ||
The concentration of iPMPA detected in Sinapis alba increased until day 28 (Fig. 3). Degradation products of sarin are also bound tightly by soils with a high organic content, and iPMPA soil spiking, following the same method as before, did not result in any recovery of iPMPA. However, by ethanol extraction of Sinapis alba, we were able to detect iPMPA out to day 28, potentially providing evidence of sarin use that would also be unobtainable using conventional procedures.
 | ||
| Fig. 3 Concentration of iPMPA in Sinapis alba. Results for each time point represent an average of analyses of four plants. | ||
The new approach for detecting nerve agent residues in the environment will aid the OPCW in their challenge to eliminate chemical weapons. The use of a localised sample and a straightforward extraction procedure also increases the likelihood of discovering CWA use. The ability of plants such as Sinapis alba to absorb nerve agents and their marker compounds protects against the removal of CW evidence, as CWAs can leach from soil over time, and suggests that green manures might be used to remediate nerve agent-contaminated sites.
Acknowledgements
The authors thank the British Ministry of Defence for funding. © Crown Copyright 2012. Published with permission of the Defence Science and Technology Laboratory on behalf of the Controller of Her Majesty's Stationary Office.References
- D. Evison, D. Hinsley and P. Rice, Br. Med. J., 2002, 324, 332 CrossRef CAS.
- R. M. Black and R. W. Read, Toxin Rev., 2007, 26, 275–298 CrossRef CAS.
- C. M. Timperley, Highly-Toxic Fluorine Compounds, in Fluorine Chemistry at the Millennium: Fascinated by Fluorine, ed. R. E. Banks, Elsevier, Oxford UK, 2000, ch. 29, pp. 499–537 Search PubMed.
- Convention on the Prohibition of the Development, Production, Stockpiling and Use of Chemical Weapons and on Their Destruction (Chemical Weapons Convention), Organisation for the Prohibition of Chemical Weapons (OPCW), The Hague, 1997 Search PubMed.
- R. M. Black, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2010, 878, 1207–1215 CrossRef CAS.
- A Strong Britain in an Age of Uncertainty: The National Security Strategy, presented to Parliament by the British Prime Minister by Command of Her Majesty, UK Stationary Office, October 2010, ISBN 9780101795326 Search PubMed.
- Sustaining US Global Leadership: Priorities for 21st Century Defense, Version 10.0, US Department of Defense, January 2012 Search PubMed.
- A. J. Bell, J. Murrell, C. M. Timperley and P. Watts, J. Am. Soc. Mass Spectrom., 2001, 12, 902–910 CrossRef CAS.
- C. M. Timperley, M. Bird, I. Holden and R. M. Black, J. Chem. Soc., Perkin Trans. 1, 2001, 26–30 RSC.
- R. M. Black and B. Muir, J. Chromatogr., A, 2003, 1000, 253–281 CrossRef CAS.
- A. F. Kingery and H. E. Allen, Toxicol. Environ. Chem., 1995, 47, 155–184 CrossRef CAS.
- N. B. Munro, S. S. Talmage, G. D. Griffin, L. C. Waters, A. P. Watson, J. F. King and V. Hauschild, Environ. Health Perspect., 1999, 107, 933–974 CrossRef CAS.
- M. W. I. Schmidt, M. S. Torn, S. Abiven, T. Dittmar, G. Guggenberger, I. A. Janssens, M. Kleber, I. Kögel-Knabner, J. Lehmann, D. A. C. Manning, P. Nannipieri, D. P. Rasse, S. Weiner and S. E. Trumbore, Nature, 2011, 478, 49–56 CrossRef CAS.
- J. L. Hambrook, D. J. Howells and D. Utley, Pestic. Sci., 1971, 2, 172–175 CrossRef CAS.
- L. E. Sverdrup, P. H. Krogh, T. Nielsen, C. Kjaer and J. Stenersen, Chemosphere, 2003, 53, 993–1003 CrossRef CAS.
- H. Winter, Sinapis in Wild Crop Relatives: Genomic and Breeding Resources, ed. C. Kole, Springer-Verlag, Berlin, 2011 Search PubMed.
- C. Montauban, A. Bégos and B. Bellier, Anal. Chem., 2004, 76, 2791–2797 CrossRef CAS.
- Y. C. Yang, L. L. Szafraniec, W. T. Beaudry, D. K. Rohrbaugh, L. R. Procell and J. B. Samuel, J. Org. Chem., 1996, 61, 8407–8413 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |