 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiomimetic membrane platform containing hERG potassium channel and its application to drug screening†
Ahu
Arslan Yildiz
*a,
CongBao
Kang
b and
Eva-Kathrin
Sinner
c
aInstitute of Materials Research and Engineering (IMRE), 3 Research Link, 117602, Singapore. E-mail: arslana@imre.a-star.edu.sg; Fax: +65 6774 1042; Tel: +65 6874 7166
bExperimental Therapeutics Centre (ETC), 31 Biopolis Way Nanos Level 3, 138669, Singapore
cUniversity of Natural Resources and Applied Life Sciences (BOKU), Muthgasse 11, 1190 Vienna, Austria
First published on 21st January 2013
Abstract
The hERG (human ether-à-go-go-related gene) potassium channel has been extensively studied by both academia and industry because of its relation to inherited or drug-induced long QT syndrome (LQTS). Unpredicted hERG and drug interaction affecting channel activity is of main concern for drug discovery. Although there are several methods to test hERG and drug interaction, it is still necessary to develop some efficient and economic ways to probe hERG and drug interactions. To contribute this aim, we have developed a biomimetic lipid membrane platform into which the hERG channel can be folded. Expression and integration of the hERG channel was achieved using a cell-free (CF) expression system. The folding of hERG in the biomimetic membrane system was investigated using Surface Plasmon Enhanced Fluorescence Spectroscopy (SPFS) and Imaging Surface Plasmon Resonance (iSPR). In addition, the hERG channel folded into our biomimetic membrane platform was used for probing the channel and drug interactions through fluorescence polarization (FP) assay. Our results suggest that the biomimetic system employed is capable of detecting the interaction between hERG and different channel blockers at varied concentrations. We believe that our current approach could be applied to other membrane proteins for drug screening or other protein-related interactions.
Introduction
Membrane proteins (MPs) are important key regulators in many biological processes such as ion transportation, generation of energy or transduction of signals across cell membranes. It is estimated that there are over 30% of the genome encoding MPs. The number of MPs being well studied is still limited due to many challenges including the need to obtain a large amount of target protein and selection of a suitable membrane-mimicking system to sustain the structure and function of a MP. A major limitation is inadequate expression systems and insufficient amounts of protein samples. Low expression efficiency, toxicity to the host cell, aggregation and folding problems are common difficulties when in vivo expression systems are used.1 The cell-free (CF) protein expression method is a powerful alternative for protein production2–5 and has been used more recently to express many MPs to overcome all of these difficulties.6,7 The CF system brings many advantages to this field and enables the addition of necessary components such as detergents or lipid membrane at any time. Addition of membrane-mimicking systems such as detergent micelles or lipids enables the folding of MPs into those membrane-mimicking media. Previously it has been reported that mild detergents such as digitonin, LMPG and Brij derivatives are suitable for soluble and functional membrane protein production.8–10 Although detergents provide a suitable system for MP folding, tolerance of the CF system to detergents is limited. An excess amount of detergents could cause denaturation, disruption of function or even inhibition of protein production.In this work we intended to set up a system for expression, purification and characterization of MPs by combining a CF approach with an artificial membrane platform (Fig. 1) instead of detergent micelles. An artificial membrane mimic not only overcomes expression limitations but also provides a robust experimental platform for many surface-science-based characterization techniques.11–13 Recently, supported membrane platforms have become involved in many membrane-related biological processes.14,15 They have become highly promising tool for the development of membrane-based biosensors and for pharmacological studies of drug screening.16,17
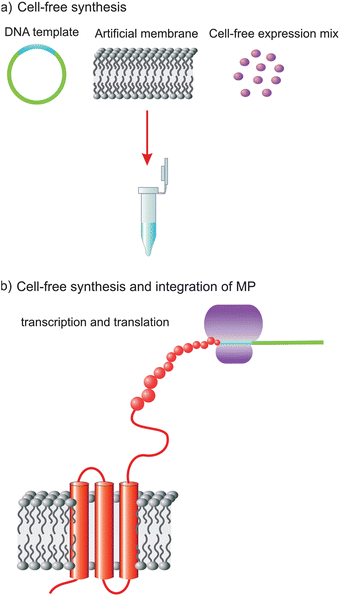 | ||
| Fig. 1 Schematic representation of CF expression and integration of a MP in an artificial membrane platform: (a) CF expression reaction mix, containing CF mixture, DNA of target protein (hERG) and artificial membrane platform; and (b) MP expressed and inserted in biomimetic membrane platform. | ||
We chose the hERG (human ether-à-go-go-related gene) potassium channel in this study because it is one of the most important targets in drug discovery. The hERG channel is a member of the voltage-gated potassium channel family. hERG channels are generally involved in action potential repolarization,18 and gain- or loss-of-function mutation can lead to sudden death.19 Due to its importance, hERG–drug interactions have been studied extensively.20–27 The hERG contains an N-terminal domain, six transmembrane segments and a C-terminal domain. The gating of the hERG channel is regulated through its N- and C-termini. To reduce the complexity, we made a construct containing only the six transmembrane segments and expressed this construct using a CF system. In this context we used the hERG-containing biomimetic platform to probe its interaction with drugs. Herein, we describe the details for the preparation, characterization via immunoblotting, Surface Plasmon Enhanced Fluorescence Spectroscopy (SPFS), Imaging Surface Plasmon Resonance (iSPR) and Fluorescence Polarization (FP) and possible application of the biomimetic membrane platform for drug screening.
Materials and methods
Materials
1-Myristoyl-2-hydroxy-sn-glycero-3-phospho-(1′-rac-glycerol) (LMPG) and 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine (DMPE) were purchased from Avanti Polar Lipids (USA). Brij S20 (homolog of Brij78), α-laminin peptide (P19), Astemizole, E4031, haloperidol and thioridazine were purchased from Sigma Aldrich (Germany). N-Hydroxysuccinimide (NHS), L-α-phosphatidylcholine from soybean (PC) and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) were purchased from Fluka (Germany). Dulbecco's phosphate buffered saline 1×, pH 7.4 (PBS) was purchased from GIBCO/Invitrogen. All reagents were used without further purification.Escherichia coli T7 S30 Extract system for Circular DNA was purchased from Promega. BCA Protein Assay Kit was obtained from Pierce Thermo Scientific (USA). All Western blot reagents and the Predictor hERG Fluorescence Polarization Assay Kit were obtained from Invitrogen. Penta·His Ab (BSA-free) mouse monoclonal IgG 1°Ab and Penta·His Alexa Fluor 647 conjugated 1°Ab were purchased from Qiagen.
Cloning and strain information
The cDNA encoding the six transmembrane segments (TMD) containing residues 403 to 666 of hERG was cloned into the NdeI and XhoI sites of pET16b. The insertion was confirmed by DNA sequencing. A 6× histidine tag was cloned at the N-terminus of TMD to aid protein purification and detection by Western blot analysis. We still refer to TMD as the hERG channel in the following text.Cell-free expression of hERG ion channel
E. coli T7 S30 Extract system for Circular DNA was used for CF expression of target protein according to the previously described protocol.28 Proteins were produced in either a tethered bilayer lipid membrane (tBLM) platform or detergent solution. Self-assembly of the tethered bilayer lipid membrane (tBLM) platform was performed step by step as described previously.28,29 The CF mixture containing plasmid DNA was added to the tBLM system upon lipid membrane formation. Detergents were supplied during the CF reaction in varied concentrations: 1-myristoyl-2-hydroxy-sn-glycero-3-phospho-(1′-rac-glycerol) and BrijS20 from 0.01 to 0.5%. Protein concentrations were determined using BCA Protein Assay Kit.Western blot analysis
CF-expressed proteins were separated on SDS-PAGE gel after mixing the protein samples with SDS sample buffer containing reducing agent. Proteins were transferred to PVDF membranes and further treatment was carried out using Western Breeze Chemiluminescent Kit (Invitrogen). Anti-His primary antibody, Penta·His Ab (BSA-free) mouse monoclonal IgG 1°Ab was used for recognition of the target protein. Blots were visualized under LAS 3000 Imaging System (Fuji) using a chemiluminescence detection method.Detection of hERG expression and integration via SPFS and iSPR
CF expression of the hERG TMD was performed directly on the surface of the freshly prepared tBLM platform according to a previously reported protocol.28 The CF expression reaction was carried out by adding plasmid DNA to the T7 S30 extract according to the manual of the expression kit (Promega) and the reaction mixture was kept at 37 °C for 2 h. All reactions were carried out step by step in the flow-cell of the SPR setup. Prior to completion of the CF reaction, the biomimetic membrane platform containing hERG TMD was treated with 0.5% blocking solution (Roche) to prevent non-specific binding of antibodies. Furthermore, fluorescence labeling of CF-expressed hERG was performed by applying Penta·His Alexa Fluor 647 conjugated 1°Ab for SPFS measurements. PBS buffer was used to rinse the samples between each step.For the SPFS measurements the SPR setup made in-house (MPIP-Mainz) and a 50 μl flow cell with Kretschmann configuration was used. A Nanofilm-EP3 (Accurion, Göttingen, Germany) setup with a BK7 flow-cell and prism in Kretschmann configuration was used for iSPR measurements. Data were collected from at least three independent measurements with baseline subtraction.
Drug binding analysis
Drug binding was determined using a Predictor hERG Fluorescence Polarization Assay Kit. Drug binding assay was carried out as explained in the assay protocol using astemizole, E4031, haloperidol or thioridazine as an inhibitor. For drug binding assay free-standing PC bilayers in vesicular configuration (50 nm) were used to ease the applicability and measurement. Vesicular membranes were added to the CF expression mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 ratio) prior to synthesis. CF lysate was loaded onto an illustra NAP-10 column (GE Healthcare) to separate CF-expressed hERG from CF mixture. Amicon Ultra concentration filters (Millipore) were used to concentrate the final protein sample. The concentrated hERG sample was used for drug binding analysis immediately.
:10 ratio) prior to synthesis. CF lysate was loaded onto an illustra NAP-10 column (GE Healthcare) to separate CF-expressed hERG from CF mixture. Amicon Ultra concentration filters (Millipore) were used to concentrate the final protein sample. The concentrated hERG sample was used for drug binding analysis immediately.
Dose responses and IC50 values were calculated from three independent measurements. Data were analyzed and dose response curves were fitted using Origin Pro8.0 software in DoseResp fitting mode.
Results and discussion
Optimization of CF expression conditions
Previous studies indicated that detergents or lipid were necessary for the folding of a membrane protein in a CF system. Although detergent micelles have been successfully appointed to MP synthesis in CF systems, different proteins may prefer different detergents due to the nature of a target protein and possible inhibition of protein synthesis by the presence of the detergents. A lipid membrane system is a promising ‘nature-like’ system because of its close structure to the cell membrane and minor effect on the CF system. A comparative study was carried out by using both a detergent system and lipid membrane platform to obtain effective expression conditions. To evaluate the effect of different systems on the yield of the hERG channel, CF expression mixtures containing varied concentrations of PC, LMPG and BrijS20 (Fig. 2a–c, respectively) were analyzed by Wstern blot analysis.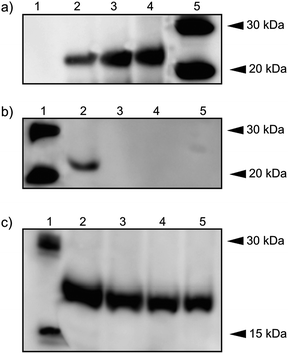 | ||
| Fig. 2 Expression of hERG using CF system. (a) hERG channel expressed by using vesicular PC lipid membrane platforms: lane 1, negative control (no DNA); lane 2, CF-expressed hERG without lipid membrane addition; lane 3, with lipid membrane addition (10%); lane 4, with lipid membrane addition (20%); lane 5, XP MagicMark™. (b) hERG channel expressed in presence of LMPG: lane 1, XP MagicMark™; lanes 2–5, CF-expressed hERG by addition of LMPG in corresponding concentrations: 0.01%, 0.1%, 0.2% and 0.5%. (c) hERG channel expressed in the presence of BrijS20: lane 1, XP MagicMark™; lanes 2–5, cell-free expressed hERG by addition of BrijS20 in corresponding concentrations: 0.01%, 0.1%, 0.2% and 0.5%. | ||
Prominent bands of hERG were detected at 24 kDa in the presence of PC, LMPG and BrijS20. The highest amount of hERG was obtained with 20% of PC lipid membrane addition. As judged by Western blot analysis, an increasing amount of PC lipid membrane resulted in an increased amount of hERG expression (Fig. 2a). This result indicates that PC lipid did not inhibit the protein synthesis and, moreover, the presence of the lipid membrane eases the folding of the hERG channel in the system. Without the addition of the membrane there is a slight expression product (Fig. 2a, lane 2), which may arise from the fact that the folding of hERG into the membrane patches exists in the CF extract. These samples were further analyzed for quantification and the findings were supported by the BCA method (see ESI, Table S1†). The highest yield of hERG with 169.3 μg ml−1 was obtained in the presence of 20% PC.
Addition of LMPG to the CF reaction mixture did not produce detectable expression bands when its concentration was above the critical micelle concentration (CMC) (Fig. 2b, lanes 3–5), while hERG expression was only observed at CMC (lane 2). With addition of BrijS20, increased concentrations of detergent resulted in decreased amount of hERG expression (Fig. 2c, lane 2–5). These results indicated that the lipid membrane platform provides a suitable environment for CF expression and insertion of hERG, while an increased amount of detergent disrupted the expression process.
SPFS and iSPR measurements
A tBLM system, consisting of P19/DMPE/PC, was assembled on a gold surface for CF expression and insertion of hERG. The artificial membrane system provided not only a nature-like environment for expression and protein folding but also a more robust platform for the study of surface-sensitive techniques. Hence, utilization of the tBLM approach eases the characterization of hERG expression and its folding in the membrane via SPFS and iSPR methods. Fig. 3b shows the SPFS signal observed for hERG obtained from CF expression. Upon expression of hERG directly in the tBLM, blocking solution and Penta·His Alexa Fluor 647 conjugated 1°Ab were allowed to flow over the surface with the hERG channel. Real-time fluorescence kinetics of Alexa Fluor 647 conjugated 1°Ab binding were monitored (Fig. 3a) and the SPFS scan was performed after PBS rinsing to remove the excess fluorophore molecule. In order to demonstrate the reliability of the measurement, the same experiment was conducted as a negative control. The magnitude of the fluorescence signal is 9 × 104 for the CF-expressed hERG sample, which was significantly higher than that of the control experiment which exhibited as a low signal as the baseline. The positive signal originating from the interaction of the hERG ion channel with Alexa Fluor 647 conjugated 1°Ab indicates that expression and insertion of the target protein to the membrane is successful (Fig. 3b).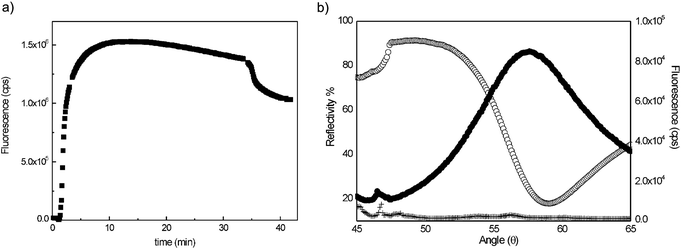 | ||
| Fig. 3 (a) Real-time fluorescence kinetic curve for Alexa Fluor 647 conjugated 1°Ab binding. (b) SPFS signal for (●) CF expression of hERG channel in biomimetic membrane platform, (+) CF reaction mixture without DNA (negative control, and (○) angular scan curve. | ||
Complementary iSPR measurements were performed to further characterize tBLM and protein insertion. For this purpose an array-type tBLM was established to provide an optical contrast and ease the visualization. Unlike the SPFS protocol, P19 assembly was achieved by μ-contact printing using 50 μm featured PDMS stamps for the formation of membrane patterns. After μ-contact printing the remaining bare gold surface was back-filled with a blocking agent 11-mercapto-1-undecanol to provide a non-adhesive surface. The thickness maps of tBLM in the absence and presence of hERG are presented in Fig. 4a and 4b respectively. The calculated thickness increase is 1.75 nm (Fig. 4c) for the regions that consist of expressed and inserted hERG. It is interesting to see such changes in the presence and absence of the hERG channel. There are flexible residues of the hERG channel based upon the X-ray structure of the potassium channel.30 These residues at the external part of hERG can be ∼1.5 nm. Therefore, the increase in thickness may arise from the insertion of hERG into the tBLM platform during CF expression.
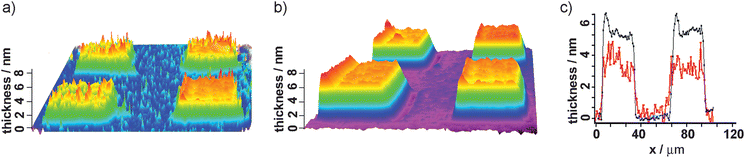 | ||
| Fig. 4 iSPR characterization for CF expression of the hERG channel in the biomimetic membrane platform; thickness map of tBLM (a) in the absence of hERG, (b) in the presence of hERG and (c) height profile of layers (red: tBLM; blue: hERG). | ||
hERG–drug interaction
Previous studies demonstrated that the sites for interaction between hERG and drugs were located at the central cavity of the channel.31 The construct made in this study was used for probing hERG and drug interactions. Fluorescence polarization (FP) assay was applied as described in the Materials and methods section. The predictor hERG tracer red molecule was used for assay development. Fig. 5a illustrates the detection strategy of drug binding assay which is based on monitoring the change in fluorescence polarization of the tracer molecule.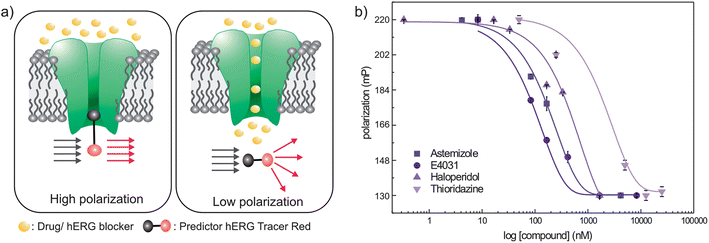 | ||
| Fig. 5 (a) Fluorescence polarization measurement mechanism, (b) dose response curves and corresponding fitting curves of cell-free-expressed hERG: (■) astemizole, (●) E4031, (▲) haloperidol and (▼) thioridazine screening. | ||
Upon CF expression of hERG in the biomimetic membrane platform, subsequent tests were performed to check the applicability of the drug binding assay. For this purpose, four well-known hERG channel blockers with a wide range of affinities were tested: astemizole, E-4031, haloperidol and thioridazine. A series of dose response curves (Fig. 5b) were obtained in the presence of the CF-expressed hERG channel, tracer red and corresponding drugs. The detection limit was optimized by inspecting varied concentrations of the drug molecules from nM to mM. As explained previously,26 non-hERG binding signals were corrected via an E-4031 titration method, and hereby the dose response curves' minima and maxima values were fixed by saturating with 30 μM E-4031.
The obtained IC50 values are given in Table 1 with Z′ > 0.88 within 2–3 h and they are comparable with reported values.26 The anomaly observed for astemizole screening can be attributed to its ability to displace with the tracer only at high concentrations.26 Although dose response curves were right-shifted compared to previously published values,21,32–35 the evaluated IC50 values fall within the acceptable range. This shifting may arise from using the TMD of hERG in this study. Nevertheless, this assay does not supply detailed information concerning the interaction of blockers with hERG or the nature and kinetics of the binding.36 It offers a versatile methodology and robust assay platform to investigate the drug binding ability of hERG for pharmacological tests. Overall it was shown that the fabricated biomimetic membrane platform containing hERG from the CF expression system could be utilized to test the interaction between hERG and its blockers over a wide range of concentrations.
| Compound | IC50/nM | Z′ |
|---|---|---|
| E-4031 | 95 | 0.88 |
| Astemizole | 180 | 0.91 |
| Haloperidol | 420 | 0.90 |
| Thioridazine | 2160 | 0.94 |
Conclusions
In conclusion, an artificial lipid membrane platform was constructed to mimic the cell-membrane for CF expression of the hERG channel. hERG in the artificial membrane showed molecular interaction with drugs. Our results show that the biomimetic membrane platform is a promising alternative approach to the reconstitution method by improving the stability of the membrane proteins and their applicability for sensing assays. The proposed method integrates the advantages of a robust platform system with the CF expression process which eliminates tedious expression and purification steps. This strategy offers a robust and rapid system for high-throughput analysis of hERG–drug interactions. Furthermore, it enables the development of pharmacological facilities to test drug–protein interactions and can be also extended to other membrane proteins.Acknowledgements
The authors would like to thank Dr Umit Hakan Yildiz for iSPR analysis and discussion. They also thank Qiwei Huang for plasmid construction. This work was supported by A*STAR JCO Grant (10/03/FG/06/06) of Singapore. C.K. appreciates the support from A*STAR Investigatorship.References
- M. Bannwarth and G. E. Schulz, Biochim. Biophys. Acta, 2003, 1610, 35 Search PubMed.
- M. W. Nirenberg and J. H. Matthaei, Proc. Natl. Acad. Sci. U. S. A., 1961, 47, 1588 CrossRef CAS.
- G. Zubay, Annu. Rev. Genet., 1973, 7, 267 CrossRef CAS.
- A. S. Spirin, Cell-Free Translation Systems, Springer, New York, 2002 Search PubMed.
- J. R. Swartz, Nat. Biotechnol., 2009, 27, 731 CrossRef CAS.
- J. U. Bowie, Nature, 2005, 438, 581 CrossRef CAS.
- F. Katzen, G. Chang and W. Kudlicki, Trends Biotechnol., 2005, 23, 150 CrossRef CAS.
- C. Klammt, D. Schwarz, K. Fendler, W. Haase, V. Dotsch and F. Bernhard, FEBS J., 2005, 272, 6024 CrossRef CAS.
- D. Schwarz, C. Klammt, A. Koglin, F. Lohr, B. W. Schneider, V. Dotsch and F. Bernhard, Methods, 2007, 41, 355 CrossRef CAS.
- C. Tian, C. G. Vanoye, C. Kang, R. C. Welch, H. J. Kim, A. L. George Jr and C. R. Sanders, Biochemistry, 2007, 46, 11459 CrossRef CAS.
- A. A. Brian and H. M. McConnell, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 6159 CrossRef CAS.
- E. Sackmann, Science, 1996, 271, 43 CAS.
- C. R. Mateo, J. Gomez, J. Villalain and J. M. Gonzales Ros, Protein–Lipid Interactions, Springer-Verlag, Berlin, 2008 Search PubMed.
- W. Knoll, I. Köper, R. Naumann and E. K. Sinner, Electrochim. Acta, 2008, 53, 6680 CrossRef CAS.
- S. Demarche, K. Sugihara, T. Zambelli, L. Tiefenauer and J. Vörös, Analyst, 2011, 136, 1077 RSC.
- M. A. Cooper, Nat. Rev. Drug Discovery, 2002, 1, 515 CrossRef CAS.
- Y. Fang, L. Y. Hong, B. Webb and J. Lahiri, MRS Bull., 2006, 31, 541 CrossRef CAS.
- M. C. Sanguinetti, C. Jiang, M. E. Curran and M. T. Keating, Cell, 1995, 81, 299 CrossRef CAS.
- M. C. Sanguinetti and M. Tristani-Firouzi, Nature, 2006, 440, 463 CrossRef CAS.
- K. Finlayson, L. Turnbull, C. T. January, J. Sharkey and J. S. Kelly, Eur. J. Pharmacol., 2001, 430, 147 CrossRef CAS.
- G. J. Diaz, K. Daniell, S. T. Leitza, R. L. Martin, Z. Su, J. S. McDermott, B. F. Cox and G. A. Gintant, J. Pharmacol. Toxicol. Methods, 2004, 50, 187 CrossRef CAS.
- P. J. Chiu, K. F. Marcoe, S. E. Bounds, C. H. Lin, J. J. Feng, A. Lin, F. C. Cheng, W. J. Crumb and R. Mitchell, J. Pharmacol. Sci., 2004, 95, 311 CrossRef CAS.
- A. E. Dubin, N. Nasser, J. Rohrbacher, A. N. Hermans, R. Marrannes, C. Grantham, K. Van Rossem, M. Cik, S. R. Chaplan, D. Gallacher, J. Xu, A. Guia, N. G. Byrne and C. Mathes, J. Biomol. Screening, 2005, 10, 168 CrossRef CAS.
- S. Whitebread, J. Hamon, D. Bojanic and L. Urban, Drug Discovery Today, 2005, 10, 1421 CrossRef CAS.
- M. H. Bridgland-Taylor, A. C. Hargreaves, A. Easter, A. Orme, D. C. Henthorn, M. Ding, A. M. Davis, B. G. Small, C. G. Heapy, N. Abi-Gerges, F. Persson, I. Jacobson, M. Sullivan, N. Albertson, T. G. Hammond, E. Sullivan, J. P. Valentin and C. E. Pollard, J. Pharmacol. Toxicol. Methods, 2006, 54, 189 CrossRef CAS.
- D. R. Piper, S. R. Duff, H. C. Eliason, J. Frazee, E. A. Frey, M. Fuerstenau-Sharp, C. Jachec, B. D. Marks, B. A. Pollok, M. S. Shekhani, D. V. Thompson, P. Whitney, K. W. Vogel and S. D. Hess, Assay Drug Dev. Technol., 2008, 6, 213 CrossRef CAS.
- L. Becucci, M. V. Carbone, T. Biagiotti, M. DAmico, M. Olivotto and R. Guidelli, J. Phys. Chem. B, 2008, 112(4), 1315 CrossRef CAS.
- A. Arslan-Yildiz, W. Knoll, R. B. Gennis and E. K. Sinner, Anal. Biochem., 2012, 423, 39 CrossRef.
- A. Arslan-Yildiz, U. H. Yildiz, B. Liedberg and E. K. Sinner, Colloids Surf., B, 2013, 103, 510–516 CrossRef.
- S. B. Long, X. Tao, E. B. Campbell and R. MacKinnon, Nature, 2007, 450, 376 CrossRef CAS.
- M. C. Sanguinetti and J. S. Mitcheson, Trends Pharmacol. Sci., 2005, 26, 119–124 CrossRef CAS.
- C. Chouabe, M. D. Drici, G. Romey, J. Barhanin and M. Lazdunski, Mol. Pharmacol., 1998, 54, 695 CAS.
- H. Tie, B. D. Walker, S. M. Valenzuela, S. N. Breit and T. J. Campbell, Lancet, 2000, 355, 1825 CrossRef CAS.
- S. Kongsamut, J. Kang, X. L. Chen, J. Roehr and D. Rampe, Eur. J. Pharmacol., 2002, 450, 37 CrossRef CAS.
- J. Wang, K. Della Penna, H. Wang, J. Karczewski, T. M. Connoly, K. S. Koblan, P. B. Bennett and J. J. Salata, Am. J. Physiol.: Heart Circ. Physiol., 2003, 284, H256 CAS.
- B. T. Priest, I. M. Bell and M. L. Garcia, Channels, 2008, 2, 87 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3an36159d |
| This journal is © The Royal Society of Chemistry 2013 |