 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Separation of metalloproteins using a novel metal ion contaminant sweeping technique and detection of protein-bound copper by a metal ion probe in polyacrylamide gel electrophoresis: distribution of copper in human serum†
Shingo Saito*a, Mitsuyoshi Kawashimaa, Hiroki Ohshimaa, Kazuki Enomotoa, Makoto Satob, Hajime Yoshimurab, Keitaro Yoshimotoc, Mizuo Maedad and Masami Shibukawaa
aGraduate School of Science and Engineering, Saitama University, 255 Shimo-Okubo, Sakura-ku, Saitama 338-8570, Japan. E-mail: shingo@apc.saitama-u.ac.jp; Tel: +81 488583559
bSagamihara R&D Center, Shino-Test Corporation, 2-29-14, Oonodai, Minami-ku, Sagmihara-shi, Kanagawa 252-0331, Japan
cGraduate School of College of Arts and Sciences, University of Tokyo, 3-8-1 Komaba, Meguro-ku, Tokyo 153-8902, Japan
dBioengineering Laboratory, RIKEN, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan
First published on 31st July 2013
Abstract
A polyacrylamide gel electrophoresis (PAGE)-based method has been developed, consisting of two types of gel electrophoresis, to obtain an accurate distribution of protein-bound metal ions in biological samples. First, proteins are separated by PAGE without the uptake of contaminant metal ions in the separation field and dissociation of metal ions from the proteins. This is followed by another PAGE for the separation and detection of protein-bound metal ions in small volume samples with high sensitivity in the ppt range using a fluorescent metal probe. The former is a new technique using blue-native (BN) PAGE to electrophoretically sweep all metal contaminants by employing two kinds of chelating agents. These agents form complexes with contaminants in the gel and the separation buffer solution, which migrate towards opposite pole directions, thus lowering the contaminants to below the ppt level during separation. This is termed “Metal Ion Contaminant Sweeping BN-PAGE (MICS-BN-PAGE)”. After the separation of proteins under these first metal-free conditions, the metal ions in the gel fractions are eluted, followed by derivatization of copper ions into the metal probe complexes to be separated and determined by fluorescence detection in the second PAGE. In this PAGE-based method, the copper ions bound to ceruloplasmin and superoxide dismutase were quantitatively determined, in addition to the exchangeable albumin-bound copper ions. This system successfully provided distribution maps of protein–copper in human serum. The precise distribution of copper in human serum was investigated, and found to be different from that which is widely accepted.
Introduction
The identification and detection of metalloproteins has recently attracted a lot of interest, since knowing which proteins bind to which metal ions in biological samples is key to understanding many biological processes involving metal ions.1–6 Various methods that enable the detection of metalloproteins have been proposed:7–10 high-performance liquid chromatography (HPLC),11,12 capillary electrophoresis (CE)13–16 and polyacrylamide gel electrophoresis (PAGE)-based techniques17–23 combined with instrumental elemental analysis methods, such as inductively coupled plasma atomic emission spectroscopy (ICP-AES), ICP-mass spectroscopy (MS), and radioautography. While these methods are very useful for investigating the distribution of metal ions in various biological samples, they do present some difficulties.HPLC-based methods24–26 are disadvantageous in that the interaction between the proteins and the hydrophobic stationary phase frequently causes denaturing of the proteins, which leads to dissociation of the metal ions. While size-exclusion HPLC (SEC) results in less denaturing of proteins due to weak interactions between proteins and the stationary phase, the resolution between the proteins is insufficient, frequently resulting in co-elution of various proteins.26–28 The CE-based method, which allows high resolution among proteins, has rather poor detection sensitivity for metal ions due to an innate and very small injection volume. Several research groups have developed PAGE-based techniques combined with laser-ablation (LA)-ICP-MS.29–32 Reasonable separation efficiencies have been obtained using these methods (with the typical number of theoretical plates for albumin in SEC, ion-exchange LC, CE and PAGE being 200,33 300,25 2700 (ref. 13) and 1200 (in this study)), with some suppression of the dissociation of metal ions from metalloproteins having been reported using the blue native (BN)-PAGE technique.30,31 However, it has been pointed out that LA-ICP-MS has poor sensitivity and irreproducibility due to the gel drying procedure and the heterogeneous surface of the gel.29,31,32 In terms of sensitivity, while the metal concentration of minor metalloproteins in biological samples is in the ppb range or lower (major metalloproteins have metal ions near the ppm level), a metal detection method at the ppt to the sub-ppb level in a small volume (∼100 μL) is required due to dilution of the samples during separation and/or fractionation in the presence of large excesses of other proteins.
In addition to these difficulties, a pitfall of all of HPLC, CE, and PAGE-based separation methods is that of contaminant metal ions.30,32 The existence of contaminant metal ions in analytical systems causes some serious problems. First, their presence results in high background noise for metal detection, namely, a high blank signal, which is usually on sub-ppb levels or higher.2 Due to the overlapping of this background noise with analyte signals, sensitive detection below the lower ppb range is impossible, and the concentrations of minor metalloproteins in biological samples are frequently in the low ppb range or less. Second, contaminant metal ions could accelerate metal-exchange processes with metalloproteins during separation, thereby resulting in a low estimate of the content of protein-bound metal ions. Third, some free proteins would bind to contaminant metal ions during migration, that is, uptake of the metal ions by apo-proteins would occur, leading to false positive detection and overestimation. These problems ensure that accurate determination of the distributions of protein-bound metal ions is difficult, particularly when determining trace metalloproteins. The amount of human serum albumin-bound copper (HSA–Cu) in human serum, which is reported to be an exchangeable metal species in human serum, has various values (3–20% total copper), depending on the analytical methods34 (vide infra).
Here, we present a set of two novel PAGE-based techniques with high sensitivity for metal ions in small volume samples, and no contamination was found during protein separation. One technique is a determination method using PAGE for metal ions (particularly copper ions) in the ppt range in ∼100 μL samples eluted from the gel fraction. This method is low-cost, robust, high-throughput35 and maintenance-free. The other technique is a contaminant metal-free BN-PAGE system, which provides protein separation without dissociation of the protein-bound metal ions and uptake of contaminant metals. Combined use of the two techniques successfully produced PAGE-based protein–copper maps to provide more accurate metal distribution information than previously reported. The proteins in the biological samples were separated using BN-PAGE with no contaminant metal ions, followed by elution of the metal ions by acid from cut gel fractions at 2.5 mm intervals. The metal ions in the eluted solution that included proteins were derivatized with fluorescent probes to yield fluorescent complexes. Thereafter, the metal complex solutions were re-concentrated and separated from metal ions and free emissive probes by the second PAGE (see Fig. S1†).
The distribution of copper ions in human serum is very important since copper ions are highly toxic36 and are strongly associated with metabolic disorders of copper such as Wilson disease,37 Menkes disease, aceruloplasminemia, and cancer,38 as well as neurodegenerative disorders such as Parkinson's disease (PD)39,40 and Alzheimer disease (AD).41,42 Copper distribution has been investigated by many researchers using various methods: ICP-MS after filtration,26 HPLC-ICP-MS,34,43 solvent extraction,44 GPC-AAS38 and isotope dilution.45 Summarizing these reports,46 the widely accepted populations of copper in human serum are 65–70% as ceruloplasmin-bound (Cp–Cu), 3–18% as human serum albumin-bound copper (HSA–Cu), and 0–9% as transcuprein-bound copper (Tc–Cu). Since the concentrations of the non-ceruloplasmin copper, which is termed as loosely bound Cu, exchangeable Cu (Ex Cu), or free Cu, are much lower than those of Cp–Cu, the determination of Ex Cu is still challenging. It has been suggested that most non-Cp–Cu binds to HSA in serum,47 and while transcuprein (Tc) is also a copper binding protein in plasma, most Tc seems to be trapped in clots to be removed from serum.48 However, there have been no reports on the quantitative determination of non-Cp–Cu complexes, including the HSA–Cu complex in serum due to dissociation of Cu ions from the HSA complex during separation. That is, there has been no direct evidence for the existence of HSA–Cu in human serum so far. In this study, we proposed a new method, Metal Ion Contaminant Sweeping-BN-PAGE/metal detection PAGE, to investigate the distribution of copper ions in human serum more accurately by controlling the dissociation of Ex-Cu (HSA–Cu).
Experimental
Chemicals
The fluorescence metal probe (FTC-ABDOTA, see Fig. 1 for the chemical structure) for metal detection PAGE was synthesized as reported previously.49 An appropriate amount of powder probe was dissolved in ultrapure water (produced by DirectQ UV, Nihon Millipore, Tokyo, >18.2 MΩ cm) to form a 2 × 10−3 M stock solution. If not otherwise specified, all reagents employed in this study were of analytical or electrophoresis grade. For gel casting, monomer stock solutions were prepared by dissolving appropriate amounts of acrylamide (AA) (Tokyo Kasei, Tokyo, Japan; >98% purity) and N,N′-methylenebisacrylamide (Bis) (Wako Pure Chemicals Industries, Ltd., Tokyo, Japan; 99% purity) in ultrapure water to a concentration of 60%T and 2.7%C, respectively. Solutions of 10% ammonium persulfate (APS, Kishida Chemical Co. Ltd., Japan) and N,N,N′,N′-tetramethylethylenediamine (TEMED, Acros Organics, Geel, Belgium) used for acceleration of polymerization were prepared at the time of use. Stock solutions of tris(hydroxymethyl)aminomethane (Tris)–HCl and Tris–glycine pH buffer were prepared by dissolving appropriate amounts of Tris and glycine powder (Sigma-Aldrich, >99% purity) in ultrapure water with the addition of ultrapure concentrated hydrogen chloride solution (20% solution, Tama Pure Chemicals, Japan) to the desired pH. Chelating agents for the MICS technique, disodium ethylenediamine-N,N,N′,N′-tetraacetate dehydrate (EDTA, >99.5% purity), N,N,N′,N′-tetrakis(2-pyridiylmethyl)ethylenediamine (TPEN, >97% purity) and trans-1,2-diaminocyclohexane-N,N,N′,N′-tetraacetic acid monohydrate (CyDTA) were purchased from Dojindo (Kumamoto, Japan). Proteins (HSA (>95% purity), superoxide dismutase (SOD) from bovine erythrocytes (Cu/Zn type) purchased from Wako Pure Chemical Industries Ltd., human Cp, and metallothionein (MT) type I from rabbit liver purchased from Sigma) were dissolved in pure water to appropriate concentrations. All protein and serum sample solutions were stored at −40 °C to prevent denaturation of the proteins.45 Bromophenol blue (BPB, 0.1 w/v%, Wako Pure Chemical Industries Ltd.), Orange G (0.1 w/v%, Tokyo Kasei) and molecular weight protein standard (Precision Plus Blue Standard, Bio-Rad Laboratories) were used as markers for PAGE. Standard metal ion solutions (Fe, Cu, Zn, Ni, Co, Mn, Cd and Hg) at 1000 ppm were purchased from Wako Pure Chemical Industries Ltd. Human blood samples (n = 10; healthy individuals) were collected in a vacuum blood collection tube, Venoject VP-P070K30 (Terumo, Tokyo, Japan). After a standing time of 3 h, the samples were centrifuged for 20 min (3000 rpm, RCF 2500g). Human serum (the supernatant liquid) was collected and stored at −40 °C to prevent denaturation.![Typical electrophoretogram using the metal probes in PAGE. Stacking gel, 16.5%T/2.7%C AA/Bis, Tris–HCl (94 mM, pH 8.85); separation gel, AA/Bis (30%T, 2.7%C), Tris–glycine (94–48 mM, pH 9.4); buffer solution, Tris–glycine (6.3–48 mM, pH 8.6); applied current constant of 30 mA; temperature constant of 4 °C; migration for 2.5 h. Sample (10 μL), [M2+] = 1.0 × 10−6 M, [L] = 1.0 × 10−5 M, [Tris–HCl] = 75 mM (pH 8.8), [glycerol] = 2.5%v/v.](/image/article/2013/AN/c3an01107k/c3an01107k-f1.gif) | ||
| Fig. 1 Typical electrophoretogram using the metal probes in PAGE. Stacking gel, 16.5%T/2.7%C AA/Bis, Tris–HCl (94 mM, pH 8.85); separation gel, AA/Bis (30%T, 2.7%C), Tris–glycine (94–48 mM, pH 9.4); buffer solution, Tris–glycine (6.3–48 mM, pH 8.6); applied current constant of 30 mA; temperature constant of 4 °C; migration for 2.5 h. Sample (10 μL), [M2+] = 1.0 × 10−6 M, [L] = 1.0 × 10−5 M, [Tris–HCl] = 75 mM (pH 8.8), [glycerol] = 2.5%v/v. | ||
Apparatus
A set of Protean XL slab gels (20 × 20 cm inner plate, 22.3 × 20 cm outer plate, 0.5 mm spacer) and a PowerPack HV high voltage power supply (Bio-Rad Laboratories) were employed for the PAGE experiments. For the gel images (absorbance for protein detection and fluorescence for metal detection), we use the gel documentation system of Printgraph AE-9633FXFC-U (Atto Corp., Japan) equipped with a monochrome CCD camera and photo-diode light sources, Gel Viewer FP 401B (Atto Corp., Japan; brightness, 1800 nt; color temperature, 6100 K; 10 W) for absorption images and Safe Imager (Invitrogen, Carlsbad, NM, λex = 470 nm, 30 W) for fluorescent images. The exposure time was set to 0.5–2.0 s. The gel images were analyzed by using CS Analyzer 3 software (Atto). A Typhoon 9400 model confocal laser scanning system (GE Healthcare Japan, Tokyo) was also used with a 520 nm bandpass filter and excitation by an Ar laser at 488 nm. A Perkin-Elmer Optima 5300 DV ICP-atomic emission spectrometer (AES) was used to determine the metal contamination concentrations in the reagents.Procedure
Results and discussion
Metal ion detection PAGE using a fluorescence probe
In metal detection PAGE, stacking and separation gels are employed. A discontinuous gel system is very useful because it can re-concentrate the diluted samples (∼100 μL) after extraction from small gel fractions (the volume of bands or spots are usually some tens of μL in PAGE). The well-known Laemmli50 and Ornstein–Davis buffer systems,51 which were designed for concentration and separation of proteins using Tris–HCl as the gel buffer (pH 6.8 and pH 8.8 for the stacking and separation gel, respectively) and Tris–gly as the migration buffer, were initially examined for metal–probe complex samples. Although the samples were stacked in thin bands, less than 1 mm wide based on the isotachophoresis mechanism,52 the stacking mode was not canceled after migration into the separation gel, i.e., the separation mode did not commence like that of the protein. This is due to the different stacking mechanisms between the proteins and the probe complex. For proteins, the change of mobility due to pH jump from the stacking gel (pH 6.8) to the separation gel (pH 8.8) causes a change in the order of mobility for the isotachophoretic concentration:52 −μglycine < −μprotein < −μCl in the stacking gel for concentration and −μprotein < −μglycine < −μCl in the separation gel due to the acid-dissociation of glycine (pKa2 = 9.66) for separation.30 For metal–probe complexes, it seemed that the order did not change, −μglycine < −μprobe < −μCl, at both pH 6.8 and 8.8, leading to the isotachophoretic stacking. This problem was resolved by a transient isotachophoresis technique: glycine was added to the separation gel. This caused disorder of the mobility since glycine exists ubiquitously in the separation gel so that transition from the stacking mode to the separation mode takes place. A 40-fold concentration enhancement was observed when 100 μL of the sample was injected.It was reported that it is very difficult to achieve separation of metal–probe complexes using the usual zone electrophoresis since they possess a very similar charge/size ratio among different center-metal ions.49,53–55 In fact, the mobility of the probe complex is the same when using the usual total gel concentrations (5–10%T) in the separation gel (see Fig. S2†). However, the resolution among the metal–probe complexes dramatically increased when an unusually high concentration gel (20–30%T) was employed (Fig. S2†). It is interesting to note that the separation of small molecules can be controlled by the gel concentration without any other manipulation.49,53,55 The results of the metal–probe complexes in PAGE with 30%T/2.7%C are shown in Fig. 1. The metal ions of Fe2+, Cu2+, Ni2+, Co2+ and Cd2+ were separated without dissociation during the separation process. In contrast, it is reported that a metal complex dissociates into the free metal ion and free ligand (probe) in the separation fields of CE or HPLC, resulting in the disappearance of the metal complex peaks in the electropherogram, if the complex is kinetically labile.49,53,54,56,57 Our results indicate that the metal–probe complexes detected in these experiments were kinetically inert. The metal ions were detected with direct fluorescence since the probe was specifically designed to show emission with or without complexation with heavy and paramagnetic metal ions,49,54,58 while these metal ions generally have the quenching effect of a fluorophore in a ligand. Direct fluorescence detection of heavy metal ions in separation methods is rare and no studies have taken place using PAGE to our knowledge. While two bands for one metal ion and a free probe were observed for Fe2+, Cu2+, Ni2+, and Cd2+, these appeared to be due to a structural isomer of the metal complex and contaminant calcium in the buffer, respectively, according to a previous report.49
The control of such high resolution among the metal ions (Fig. 1) may be due to several possible factors. One is the molecular sieving effect. The dependence of the resolution on the gel concentration was investigated, and the pore radius in the gel was estimated according to equations reported by Holms and Stellwagen59,60 (see Fig. S2†). Decreasing the pore size (40–101 nm) and increasing the total gel concentration (5–30%T/2.7%C) resulted in a higher resolution, while not even a slight separation was achieved with the smallest pore size with high concentration of the cross-linker (estimated pore size: 29 nm, Fig. S2e†). In addition, the mobility of the metal–probe complex was independent of the degree of cross-linking (Fig. S3†). As such, the sieving effect does not seem to work in the separation of metal complexes. Another possibility is the adsorption of the probe complexes on acrylamide residues in the gel. If adsorption should take place, the resolution would change depending on the gel concentration, and the mobility of the probe complexes would decrease with increasing gel concentration because the number of adsorption sites increases. However, the constant mobility of the probe complexes was observed at over 15%T in experiments using capillary gel electrophoresis (data not shown). Based on this fact, few adsorptions onto the residue in the gel seemed to occur. Another explanation for the separation mechanism is the effect of unusual water in the nanopores. Shibukawa et al. reported that the selectivity of various small water-soluble molecules in gel filtration chromatography depends on the amount of solutes in the freezable bound water and non-freezable water as opposed to the bulk water that functions as a stationary phase in water-swollen hydrophilic polymer gels including polyacrylamide gel, and suggested that the water differs from the usual bulk water in hydrophilic gels.61,62 It has also been reported that water in a nanotube has high viscosity and low polarity, and stabilizes the hydration structure of a protein.63 Although the detailed separation mechanism is beyond the scope of this work, the separation of small molecules in nanopores is an interesting subject.
The detection limits (DLs) in the metal detection PAGE were determined for metal ions completely separated using both the CCD camera with the transilluminator and the fluorescence imager with an Ar laser and are summarized in Table 1 (see Fig. S4 for the calibration curve and electropherogram†). Even using excitation by the transilluminator, high sensitivity for sub-ppb (nM order) was accessible and sensitivity reached the ppt range (or sub-femtomol) using a confocal laser scanning system. Such high sensitivity without employing large analytical instruments such as ICP-MS is useful for a biochemist familiar with low-cost PAGE.
| Metal ion | Detection by a CCD camera/M (ppb, amount/pg) | Detection by a laser-excitation imager/M (ppb, amount/pg) |
|---|---|---|
| a The detection limits were determined based on the 3σ of blank. Linearity was obtained from 0.1 to 100 ppb. | ||
| Fe | 3.3 × 10−9 (0.18, 1.8) | 1.9 × 10−10 (0.010, 0.10) |
| Cu | 2.6 × 10−9 (0.17, 1.7) | 7.8 × 10−11 (0.0051, 0.051) |
| Ni | 3.8 × 10−9 (0.22, 2.2) | 2.1 × 10−10 (0.012, 0.12) |
| Co | 3.2 × 10−9 (0.19, 1.9) | 1.2 × 10−10 (0.007, 0.07) |
| Cd | 7.4 × 10−9 (0.83, 8.3) | 2.2 × 10−10 (0.025, 0.25) |
The metal detection PAGE system was applied to some metalloprotein standard solutions and a standard human serum sample to confirm whether this system would work for biological samples. Solutions of Cp, SOD, HSA and MT, and a human serum sample were examined to determine the total copper and cadmium concentrations in metal-binding proteins and in a real biological sample. The results obtained are summarized in Table S1.† The values determined were almost identical to the concentrations determined by ICP-AES. These results suggest that this type of PAGE is suitable for the detection of metal ions with higher affinity to metal ions than the coexisting metal-binding proteins. These results are reasonable considering the higher affinity of DOTA towards metal ions (log KM–DOTA = 22.3, 21.3 M−1 for Cu2+, Cd2+, respectively)64 than the metalloproteins used (log KM–protein values of 16.2,65 15 for HSA–Cu, SOD–Cu and MT–Cd).66
Metal ion contaminant sweeping (MICS)-blue native (BN)-polyacrylamide gel electrophoresis (PAGE)
The metal detection PAGE (as described above) was applied as the second dimension to the sample solutions, which were extracted by 0.2 M HCl from cut gel fractions (25 μL) after conventional BN-PAGE of human serum samples.67 Since some researchers have reported that metal ions strongly binding to proteins do not dissociate during migration using BN-PAGE,30,31 it was expected that the distribution of the metal ions could be obtained by combining BN-PAGE and metal detection PAGE. The electropherogram (a protein–metal map) obtained is shown in Fig. 2. No cadmium and nickel ions were detected in the fractions, since the amounts of Cd and Ni in human serum are generally very small. Iron, cobalt and manganese ions were also not detected due to interference from the rather thick bands of the free probe since a large excess of the probe (5 × 10−6 M) had to be employed for quantitative derivatization (total Fe and Cu concentration expected was up to the μM level in the gel fraction for a human serum sample). Copper ions were successfully detected in the human serum sample. However, they were detected in many gel fractions, while the ceruloplasmin-bound copper (Cp–Cu) was detected as expected (see Fig. 2). If this measurement was accurate, the summation of the amount of Cu in each gel fraction would correspond to the total concentration of Cu in the serum. The recovery of total Cu in this method based on the summation was 643% (6.1 ppm Cu) compared to the total Cu obtained by ICP-AES (0.95 ppm Cu). Meanwhile, since the quantitative extraction of copper ions from gel fractions was confirmed by a recovery test adding copper standard solution to the gel monomer solution prior to polymerization (98 ± 9.4%, n = 6, data not shown), this BN-PAGE/metal detection PAGE system seemed to work.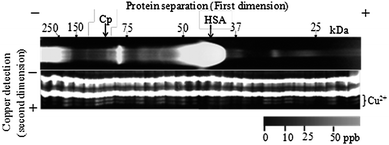 | ||
| Fig. 2 Detection of copper ion in gel fractions of conventional BN-PAGE. Sample: human serum from a healthy male, age 48. Recovery (summation of each fraction/total Cu concentration in the serum sample = 6.13 ppm/0.95 ppm) was 643%. | ||
As such, since serious contamination by Cu ions was expected for this method, each instrument and procedure was verified in detail in terms of metal contamination. The results of a comprehensive investigation into contaminant Cu and Fe (in polyacrylamide gel, migration buffer reagents, vacuum blood collection tubes employed, etc.) showed serious metal contamination from the gel in the ppb range, and migration buffer solutions at sub-ppb levels (see Table S2† and footnotes in ref. 68 and 69). According to a blank test result (metal detection PAGE of gel fractions in BN-PAGE with water sample injection, see Fig. S5†), an average Cu concentration of 7.5 ± 1.7 ppb per single fraction was found. This concentration level agrees well with the contamination levels in the reagents employed. The proteins may have been separated in the separation media including metal contamination in the ppb range by various methods previously reported, such as PAGE and LC using high concentration buffers and/or gels comprised of even analytical grade reagents. Although serious contamination might render accurate measurements of the distribution of metals impossible due to the high background, uptake of metals into proteins and acceleration of metal-exchange reactions of metalloproteins during migration or elution (see the footnote in ref. 70), this essential problem remains unresolved. In terms of the metal contamination problem, the uptake of contaminant Cu by HSA during separation was estimated on the basis of equilibrium calculations (see ESI†), which implies that sub-ppb contamination in the separation buffer solution in LC or electrophoresis provides a ppb range of uptake even under optimistic estimations. In the case of our PAGE, contaminant Cu ions possibly exist in the form of [Cu–(Tris)4]2+ in the gel and the lower (anode) buffer solution, and [Cu–(glycine)3]− in the upper (cathode) buffer solution judging from their stability constants (KCu–Tris4 = 1014.1 M−4, KCu–gly3 = 1016.96 M−4).71,72 Each complex could migrate toward the upper (cathode) and lower (anode) directions, respectively (Fig. S6a†). This implies that the contaminant Cu complexes in the buffer solutions are always supplied from both the upper and lower buffer reservoirs into the gel during migration due to the electrical charges of the contaminant metal complexes, i.e., this migration of contaminant metals would also provide a ppb level of contamination.
With the metal contamination problem in mind, we developed a new metal contamination-free PAGE, termed Metal Ion Contaminant Sweeping (MICS)-BN-PAGE (see the Experimental section for the procedure), which could completely avoid the contact of metalloproteins with contaminant metal ions during migration. The method, applied in the first dimension prior to metal detection PAGE, consists of three stages (see Fig. S6b–d†). First, we prepared stacking and separation gels including EDTA for masking the contaminant metal ions, followed by conditioning with an upper buffer solution including TPEN by applying voltage. This operation sweeps all contaminant metal ions in the stacking gel toward the lower solution, and the metal contaminants in the upper buffer solution could not migrate into the gel during conditioning because each contaminant metal (mostly transition metal ions) forms anionic [M2+/3+–EDTA4−]2−/− complexes in the gel or cationic [M2+/3+–TPEN]2+/3+ complexes in the upper buffer solution with very high thermodynamic64,73 (log KML ∼ 25) and kinetic stability.53,56,57 Second, the sample was applied with a TPEN-free upper buffer solution (buffer reagents were diluted to prevent serious contaminant metal ions) to avoid the ligand-exchange reaction of metalloproteins with TPEN with high affinity to transition metal ions.73 Applying voltage for a short time (600 V, 3 min) made the sample proteins penetrate slightly into the stacking gel (2 mm). Finally, the voltage was applied for stacking and separation after a second exchange of the upper buffer solution to that including TPEN, since the proteins in the stacking gel would not come into contact with TPEN in the upper buffer solution (the contaminant Cu in the separation gel migrates ahead of the protein bands). This MICS procedure substantially decreases the contamination level, avoiding metal-exchange and ligand-exchange processes during migration. The contamination level upon electrophoresis was estimated to be in the lower ppq level according to equilibrium calculations using stability constants (see ESI†). A blank measurement was demonstrated using MICS-BN-PAGE/metal detection PAGE (Fig. S7†). The contamination level was successfully lowered to the ppt level or lower (below the detection limit of metal detection PAGE).
An example of a protein–Cu map of human serum (for a sample obtained from a healthy human female) at low temperature (see below) by MICS-BN-PAGE/metal detection PAGE is shown in Fig. 3. No metal contamination was observed in any fraction, and the total Cu (summation of each fraction) was consistent with the total Cu determined by ICP-AES and metal detection PAGE of the serum samples (recovery 99 ± 3%, n = 10). This strongly indicated that almost all Cu bound to proteins was detected in this system. When no TPEN was added to the upper buffer solution, the recovery was 466%, which means that the penetration of the contaminant Cu from the upper buffer solution during electrophoresis was about 170% corresponding to 1.68 ppm (based on results without chelating agents, 643% recovery, vide supra) (see Fig. S8†). This revealed that contaminant Cu existed in the gel, eluted from the instruments, and actually migrated into the gel from the migration buffer solutions without the MICS technique.
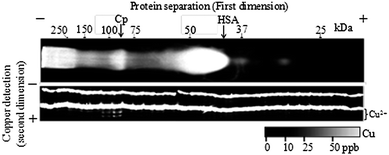 | ||
| Fig. 3 Typical copper ion mapping using MICS-BN-PAGE/copper detection PAGE for human serum (a healthy female, age 25) at low temperature (0 °C on the gel surface, −5 °C in the incubator). | ||
Protein–copper mapping of human serum: recovery of albumin-bound copper ions
The results of protein–copper mapping at low temperature (vide infra) are summarized in Table 2 (the detailed results are shown in Table S3†). Most copper ions were detected in Cp fractions but few in the HSA fraction in healthy individuals (n = 10, female and male, age 25–56), i.e., the distribution of Cu was approximately 100% Cp–Cu. This result is quite inconsistent with many previous reports, in which 4–19% of HSA-bound Cu ions were detected.26,34,38,43–45 Meanwhile, it was reported by several research groups that the detection and quantitative determination of HSA–Cu is very difficult28,34 since it is a kinetically labile metal complex.37,45| Sample | Total Cu/ppba | Total Cu found/ppb (recovery%) | Cp fraction/ppb (%) | HSA fraction/ppb (%) | 60 kDa fraction/ppb (%) | 30 kDa fraction/ppb (%) |
|---|---|---|---|---|---|---|
| a Determined by ICP-AES.b Aalto Control LEVEL II N.c Healthy individuals, age 25–56, male and female.d Denatured by four freeze–thaw cycles. All measurements at 293 K. | ||||||
| Standard human serumb | 1124 | 1082 (96.3) | 1082 (100) | n.d. (<0.26) | n.d. (<0.26) | n.d. (<0.26) |
| Human serum (n = 10)c | 764 ± 44 | 755 ± 45 (99 ± 3) | 754 ± 68 (99.9 ± 0.3) | 1 ± 4 (0.1 ± 0.5) | n.d. (<0.37) | n.d. (<0.37) |
| A human serum with the addition of 294 ppb Cu | 965 | 952 (99) | 649 (68) | 187 (20) | 116 (12) | n.d. (<0.37) |
| A human serum with 4 times refreezing cycled | 626 | 625 (99.8) | 311 (58) | 62 (12) | n.d. (<0.45) | 160 (26) |
Since we doubted whether HSA–Cu could be quantitatively determined by our mapping method, the spiking tests of Cu in standard HSA solutions incubated with the addition of Cu to form HSA–Cu were examined. HSA–Cu of 40 ± 9% was recovered from HSA fractions in the MICS-BN-PAGE/metal detection PAGE at 20 °C (see Fig. S9†). This indicates that more than half of the HSA–Cu dissociated during migration, while semi-quantitative analysis succeeded. In general, there are four possible dissociation processes for metal complexes in aqueous solution:74 acid (or base)-assisted dissociation by attacking of oxonium ion (hydroxide ion); ligand-exchange reaction by a ligand possessing coordinating ability (glycine and Tris in the buffer were the candidates in this case); metal-exchange reaction (by metal ion contaminants in this case); and spontaneous dissociation (solvolysis). In MICS-BN-PAGE, no metal-exchange process likely occurs due to the metal ion contamination level being lowered to ppt or less. We investigated where the dissociation reaction occurred in the slab gel, and we also investigated: the effects of the acid-assisted dissociation reaction by changing the pH of the gels; the ligand-exchange reaction by changing glycine to γ-aminobutyric acid and concentrations of Tris; and the spontaneous dissociation reaction by changing the temperature (data not shown). As a result, it was revealed that HSA–Cu dissociated in the stacking gel, and spontaneous dissociation predominantly occurred (no ligand-exchange and acid-assisted dissociation was observed for HSA–Cu). To decelerate the spontaneous dissociation process, the temperature of the separation gel was lowered and accurately maintained at 273 K by steeping the stacking gel in the lower buffer solution and by using the cooling core set around the gel, which was thermostatted by circulating solvent. In addition, the set of electrophoresis instruments was placed in an incubator at 268 K during migration. This thorough control of the gel temperature successfully provided the quantitative recovery of HSA–Cu without dissociation (97 ± 4%, see Fig. S9†). An intercept corresponding to 49 ppb of Cu was observed in the calibration curve, which was equivalent to the contamination Cu concentration value of the HSA standard solution, 50 ppb, determined by ICP-AES (Table S1†). When 294 ppb of Cu was added to a human serum sample, the added Cu was recovered from HSA (19.7% of total Cu) and 60 kDa (12.2% of total Cu) fractions (99% recovery, Table 2). These facts strongly indicated that quantitative determination of Ex-Cu is accessible using our system, and the distribution of Cu in the human serum samples in Table 2 was accurate. The quantitative recovery of exchangeable HSA–Cu is reported for the first time, to the best of our knowledge.
Based on our results, it was concluded that there is a high possibility that the widely accepted distributions of Cu in human serum are incorrect. The inconsistency appears to be due to the denaturing of proteins (especially Cp) in the samples, as well as the dissociation processes of protein-bound Cu ions and the uptake of contaminant Cu by proteins; the impacts of none of these processes seem to have been simultaneously estimated to date. Buckley et al. recently reported that Cp–Cu is easily denatured even at −10 °C (Cp–Cu is stable at below −60 °C), denaturing is irreversible, and cycles of freezing and refreezing also induce denaturing. Cu released from denatured Cp would bind with other proteins such as HSA. In our case, the serum samples were frozen just after centrifugation at −40 °C (see the Procedure section), and no refrozen samples were used to avoid denaturing effects. In fact, when using a deliberately denatured human serum sample via four freeze–thaw cycles, the amount of Cp–Cu clearly decreased, and the HSA–Cu concentration increased (see Table 2 and Fig. S10†). In addition, Cu ions bound with an unknown protein of 30 kDa were observed. Thus, it seems that there are multiple effects on the results reported previously. A value of 3.4% of Ex-Cu in plasma, which was the closest to our results, was reported after careful handling of samples in terms of denaturation by using the isotope dilution method.45 In this method, contamination with metal ions in the reagents would be less than in the separation methods, while the Ex-Cu content in serum should be lower than in plasma since most Tc–Cu is lost from serum into clots.48
Conclusions
Using the MICS-BN-PAGE/metal detection PAGE system, the accurate distribution of Cu in human serum was investigated by quantitative determination of exchangeable HSA–Cu. Metal detection PAGE is suitable for determination of ultratrace metal ions in many cut gel fractions after slab gel electrophoresis, including two dimensional gels, because simultaneous analysis of many samples is accessible with re-concentration of the diluted elutes.35 Since other metal ions like Fe and Hg were also detected by metal detection PAGE using other metal probes with different chelating structures (data not shown), their distribution in biological samples will be reported in our next publication. Also, this is the first report of contaminant metal-free separation media for metalloproteins with suppression of the dissociation processes, in which the protein-bound copper with slower dissociation kinetics than the exchangeable HSA–Cu can be detected. As such, MICS-BN-PAGE provides lots of potential to detect metal ions including exchangeable ones. Future experiments will focus on determination of trace metal ions binding with metalloproteins in various biological samples by using the MICS-BN-PAGE/metal detection PAGE system.Acknowledgements
This research was supported by a Grant-in-Aid for Young Scientists (B) no. 23750077 and no. 21750072 from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and by Asahi Glass Foundation, and Foundation NAGASE Science Technology Development.Notes and references
- K. J. Waldron, J. C. Rutherford, D. Ford and N. J. Robinson, Nature, 2009, 460, 823 CrossRef CAS.
- H. Haraguchi, J. Anal. At. Spectrom., 2004, 19, 5 RSC.
- A. Cvetkovic, A. L. Menon, M. P. Thorgersen, J. W. Scott, F. L. Poole II, F. E. Jenney Jr, W. A. Lancaster, J. L. Praissman, S. Shanmukh, B. J. Vaccaro, S. A. Trauger, E. Kalisiak, J. V. Apon, F. Siuzdak, S. M. Yannone, J. A. Tainer and M. W. W. Adams, Nature, 2010, 466, 779 CrossRef CAS.
- J. Szpumar, Analyst, 2005, 130, 442 RSC.
- W. Shi and M. R. Chance, Cell. Mol. Life Sci., 2008, 65, 3040 CrossRef CAS.
- I. Worms, D. F. Simon, C. S. Hassler and K. J. Wilkinson, Biochimie, 2006, 88, 1721 CrossRef CAS.
- Y. Ha, O. G. Tsay and D. G. Ghurchill, Monatsh. Chem., 2011, 142, 385 CrossRef CAS.
- J. S. Garcia, C. S. Magalhaex and M. A. Z. Arruda, Talanta, 2006, 69, 1 CrossRef CAS.
- J. Szpunar, Analyst, 2000, 125, 963 RSC.
- R. Lobinski, C. Moulin and R. Ortega, Biochimie, 2006, 88, 1591 CrossRef CAS.
- A. S. González, J. R. Encinar, A. M. C. Roldán, J. M. M. Gayón and A. S. Medel, Anal. Chem., 2008, 80, 8702 CrossRef.
- S. Trümpler, W. Lohmann, B. Meermann, W. Buscher, M. Sperling and U. Karst, Metallomics, 2009, 1, 87 RSC.
- B. Deng, P. Zhu, Y. Wang, J. Feng, X. Li, X. Xu, H. Lu and Q. Xu, Anal. Chem., 2008, 80, 5721 CrossRef CAS.
- B. Deng, X. Li, P. Zhu, X. Xu, Q. Xu and Y. Kang, Electrophoresis, 2008, 29, 1534 CrossRef CAS.
- Y. Li, X. Yin and X. Yan, Anal. Chim. Acta, 2008, 615, 105 CrossRef CAS.
- A. Nguyen and M. Moini, Anal. Chem., 2008, 80, 7169 CrossRef CAS.
- M. G. Aňorbe, J. Messerschmidt, I. Feldmann and N. Jakubowski, J. Anal. At. Spectrom., 2007, 22, 917 RSC.
- J. S. Becker, R. Lobinski and J. S. Becker, Metallomics, 2009, 1, 312 RSC.
- M. Ferre, O. V. Golyshina, A. Beloqui, P. N. Golyshin and K. N. Timmis, Nature, 2007, 445, 91 CrossRef.
- S. D. Smith, Y. She, E. A. Roberts and B. Sarkar, J. Proteome Res., 2004, 3, 834 CrossRef CAS.
- B. Kastenholz, Protein Pept. Lett., 2006, 13, 503 CrossRef CAS.
- B. Katenholz, Protein Pept. Lett., 2007, 14, 389 CrossRef.
- A. M. Sevcenco, M. W. Pinkse, H. T. Wolterbeek, P. D. Verhaert, W. R. Hagen and P. L. Hagedoorn, Metallomics, 2011, 3, 1324 RSC.
- M. Malavaolta, F. Piacenza, A. Basso, R. Giacconi, L. Costarelli, S. P. Paoli and E. Mocchegiani, Anal. Biochem., 2012, 421, 16 CrossRef.
- C. S. Muñiz, J. M. M. Gayón, J. I. G. Alonso and A. S. Muniz, J. Anal. At. Spectrom., 2001, 16, 587 RSC.
- T. Hasegawa, Y. Wakita, Y. Zhu, H. Matsuura, H. Haraguchi and T. Umemura, Bull. Chem. Soc. Jpn., 2007, 80, 503 CrossRef CAS.
- E. Z. Jahromi, W. White, Q. Wu, R. Yamdagni and J. Gailer, Metallomics, 2010, 2, 460 RSC.
- S. E. Balkhi, J. Poupon, J. Trocello, F. Massicot, F. Woimant and O. Laprévote, Anal. Chem., 2010, 82, 6904 CrossRef.
- C. C. Chéry, D. Günther, R. Cornelis, F. Vanhaecke and L. Moens, Electrophoresis, 2003, 24, 3305 CrossRef.
- M. S. Jimenez, L. Rodriguez, M. T. Gomex and J. R. Castillo, Talanta, 2010, 81, 241 CrossRef CAS.
- M. S. Jimenez, M. T. Gomez, L. Rodriguez, L. Martinez and J. R. Castillo, Anal. Bioanal. Chem., 2009, 393, 699 CrossRef CAS.
- A. Raab, B. Pioselli, C. Munro, J. Thomas-Oates and J. Feldmann, Electrophoresis, 2009, 30, 303 CrossRef CAS.
- S. A. Manley, S. Byrns, A. W. Lyon, P. Brown and J. Gailer, J. Biol. Inorg. Chem., 2009, 14, 61 CrossRef CAS.
- K. Inagaki, N. Mikuriya, S. Morita, H. Haraguchi, Y. Nakahara, M. Hattori, T. Kinosita and H. Saito, Analyst, 2000, 125, 197 RSC.
- In the gel electrophoresis instruments employed in this work, up to 160 samples can be simultaneously applied with four electrophoresis chambers and eight gel plates. Migration time per sample is less than a few minutes.
- B. A. Fowler, G. F. Nordberg, M. Nordberg and L. Friberg, Handbook on the Toxicology of Metals, Academic Press, 3rd edn, 2007, ch. 26, p. 529 Search PubMed.
- J. M. Walshe, Q. J. Med., 2012, 105, 419 CrossRef CAS.
- P. L. Wirth and M. C. Linder, JNCI, J. Natl. Cancer Inst., 1985, 75, 277 CAS.
- S. S. S. J. Ahmed and W. Santosh, PLoS One, 2010, 5, e11252 Search PubMed.
- G. N. L. Jameson, Monatsh. Chem., 2011, 142, 325 CrossRef CAS.
- G. Arena, G. Pappalardo, I. Sovago and E. Rizzarelli, Coord. Chem. Rev., 2012, 256, 3 CrossRef CAS.
- P. Faller and C. Hureau, Dalton Trans., 2009, 1080 RSC.
- L. Barrow and M. S. Tanner, Eur. J. Clin. Invest., 1988, 18, 555 CrossRef CAS.
- (a) M. M. Wintrobe, G. E. Cartwright and C. J. Gubler, J. Nutr., 1953, 29, 395 Search PubMed; (b) M. M. Wintrobe and G. E. Cartwright, Am. J. Clin. Nutr., 1964, 14, 224 Search PubMed; (c) G. W. Evans and R. E. Wiederanders, Am. J. Physiol., 1967, 213, 1183 CAS.
- W. T. Buckley, A. Richard and A. Vanderpool, Biometals, 2008, 21, 601 CrossRef CAS.
- K. D. Rainsford, R. Milanino, J. R. J. Sorenson and G. P. Velo, Copper and Zinc in Inflammatory and Degenerative Diseases, Kluwer Academic Publishers, 1998, ch. 62, p. 24 Search PubMed.
- M. E. Shils, M. Shike, A. C. Ross, B. Caballero and R. J. Cousins, Modern Nutrition In Health And Disease, Lippeincott Williams & Wilkins, 10th edn, 2005, p. 295 Search PubMed.
- A. Cabrera, E. Alonzo, E. Sauble, Y. L. Chu, M. C. Linder, D. S. Sato and A. Z. Mason, Biometals, 2008, 21, 525 CrossRef CAS.
- S. Saito, Y. Nakano, A. Hikichi, R. Suzuki, K. Yoshioto, M. Maeda, M. Aoyama and M. Shibukawa, Analyst, 2011, 136, 2697 RSC.
- U. K. Laemmli, Nature, 1970, 227, 680 CrossRef CAS.
- B. J. Davis, Ann. N. Y. Acad. Sci., 1964, 121, 404 CrossRef CAS.
- M. Urbánek, L. Křivánková and P. Boček, Electrophoresis, 2003, 24, 466 CrossRef.
- S. Saito, R. Suzuki, N. Danzaka, A. Hikichi, K. Yoshimoto, M. Maeda and M. Aoyama, Electrophoresis, 2007, 14, 2448 CrossRef.
- S. Saito, J. Shimidzu, K. Yoshimoto, M. Maeda and M. Aoyama, J. Chromatogr., A, 2007, 1140, 230 CrossRef CAS.
- S. Saito, S. Takeuchi, K. Yoshimoto, M. Maeda and M. Aoyama, Analyst, 2007, 132, 237 RSC.
- S. Saito, S. Sasamura and S. Hoshi, Analyst, 2005, 130, 659 RSC.
- S. Saito and H. Hoshino, Anal. Bioanal. Chem., 2004, 378, 1644 CrossRef CAS.
- S. Saito, Bunseki Kagaku, 2011, 60, 773 CrossRef CAS.
- D. L. Holmes and N. C. Stellwagen, Electrophoresis, 1991, 12, 253 CrossRef CAS.
- D. L. Holmes and N. C. Stellwagen, Electrophoresis, 1991, 12, 612 CrossRef CAS.
- M. Shibukawa, K. Aoyagi, R. Sakamoto and K. Oguma, J. Chromatogr., A, 1999, 832, 17 CrossRef CAS.
- M. Shibukawa, Bunseki Kagaku, 2006, 55, 149 CrossRef CAS.
- N. Kameta, H. Minamikawa, Y. Someya, H. Yui, M. Masuda and T. Shimizu, Chem.–Eur. J., 2010, 16, 4217 CrossRef CAS.
- G. Anderegg, R. Arnaud-Neu, R. Delgado, J. Felcman and K. Popov, Pure Appl. Chem., 2005, 77, 1445 CrossRef CAS.
- S. Y. Lau and B. Sarkar, J. Biol. Chem., 1971, 246, 5938 CAS.
- S. Y. Lau, J. Biol. Chem., 1971, 246, 5938 CAS.
- I. Wittig, H. P. Braun and H. Schagger, Nat. Protoc., 2006, 1, 418 CrossRef CAS.
- The contamination level of Cu from vacuum blood collection tubes was at sub-ppb, investigated by blank tests. This contamination concentration is 0.1% or lower than total Cu concentration in human serum. Therefore, the tube was not a major contamination source.
- Such ppb level contamination of metal ions was a surprise to us. However, it must be noted that contamination on the order of ppb is possible according to information from reagent suppliers. For example, a high contaminant metal content (<5 ppm for each Cu and Fe) in polyacrylamide powder (electrophoresis grade) purchased from Sigma is characterized in the specification sheet.
- In fact, the elevated baseline and saddle-shaped peaks of chromatograms frequently observed were possibly due to metal contamination, and unreasonable distributions were obtained for biological samples. For examples, see ref. 2, 18, 24 and 26.
- I. J. Arnquist and J. A. Holcombe, Spectrochim. Acta, Part B, 2012, 76, 140 CrossRef CAS.
- P. D. Angelo, E. Bottari, M. R. Festa, H. F. Nolting and N. V. J. Pavel, J. Phys. Chem., 1998, 102, 3114 Search PubMed.
- G. Anderegg, E. Hubmann, N. G. Poddr and F. Wenk, Helv. Chim. Acta, 1977, 60, 123 CrossRef CAS.
- (a) S. F. Lincoln and A. E. Merbach, Advances in Inorganic Chemistry, Academic Press, New York, 1995, vol. 42 Search PubMed; (b) A. E. Martell, Coordination Chemistry, American Chemical Society, Washington, DC, 1978, vol. 2 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3an01107k |
| This journal is © The Royal Society of Chemistry 2013 |