 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetection of an estrogen derivative in two breast cancer cell lines using a single core multimodal probe for imaging (SCoMPI) imaged by a panel of luminescent and vibrational techniques†
Sylvain Clèdeabc, François Lambertabc, Christophe Sandtd, Slavka Kascakovae, Miriam Ungerf, Etienne Hartég, Marie-Aude Plamonth, Rénette Saint-Forth, Ariane Deniset-Besseaui, Zoher Guerouiabj, Carol Hirschmuglf, Sophie Lecomteg, Alexandre Dazzii, Anne Vessièresh and Clotilde Policar*abc
aEcole Normale Supérieure, Département de chimie, 24, rue Lhomond, 75005 Paris, France. E-mail: clotilde.policar@ens.fr
bUPMC, 4, Place Jussieu, 75005 Paris, France
cLaboratoire des Biomolécules, CNRS, UMR7203, France
dSMIS, Synchrotron SOLEIL Saint-Aubin, 91192, Gif-sur-Yvette Cedex, France
eDISCO, Synchrotron SOLEIL Saint-Aubin, 91192, Gif-sur-Yvette Cedex, France
fDepartment of Physics, University of Wisconsin-Milwaukee, Milwaukee, WI 53211, USA
gCNRS-Université Bordeaux 1-IPB, CBMN UMR 5248, allée Geoffroy de St Hilaire, 33600 Pessac, France
hLaboratoire Friedel, Chimie ParisTech, CNRS, UMR7223, 11, rue Pierre et Marie Curie, 75231 Paris, France
iLaboratoire de Chimie Physique, UMR-CNRS 8000, Université Paris-Sud 11, 91405 Orsay, France
jLaboratoire PASTEUR, CNRS, UMR8640, France
First published on 2nd July 2013
Abstract
3-Methoxy-17α-ethynylestradiol or mestranol is a prodrug for ethynylestradiol and the estrogen component of some oral contraceptive formulations. We demonstrate here that a single core multimodal probe for imaging – SCoMPI – can be efficiently grafted onto mestranol allowing its tracking in two breast cancer cell lines, MDA-MB-231 and MCF-7 fixed cells. Correlative imaging studies based on luminescence (synchrotron UV spectromicroscopy, wide field and confocal fluorescence microscopies) and vibrational (AFMIR, synchrotron FTIR spectromicroscopy, synchrotron-based multiple beam FTIR imaging, confocal Raman microspectroscopy) spectroscopies were consistent with one another and showed a Golgi apparatus distribution of the SCoMPI–mestranol conjugate in both cell lines.
1 Introduction
There are nowadays many non-invasive techniques for cellular detection of tagged molecules and fluorescent tags are widely developed. Vibrational spectroscopies are attractive for bio-imaging,1–4 in the case of vibrational excitations in the IR, where no photo-bleaching is induced,5 in contrast to what is observed with organic fluorophores in the visible or UV-range. IR-probes or tags have been reported in the literature and they are mostly metal-carbonyl units that show many advantages (stability in the biological environment, intense absorption in the 1800–2200 cm−1 range which is the transparent IR window of biological media).6 They are used for immunoassay7,8 and in a few cases of cellular mapping using IR5,9−13 or Raman spectroscopy.14,15 The smaller the probe, the less modified the physico-chemical properties of the molecule and its location in biological tissues will be. This is why small IR-tags are attractive.16 Interestingly metal-carbonyl bearing specific ancillary ligands with low π* orbitals are known to be luminescent.17,18 In the case of the rhenium-centred probes, emission properties allow their use in bio-imaging.19–26Coupling IR and luminescence detection using metal-carbonyl can then be envisioned. Indeed, we have recently developed the concept of single core multimodal probe for imaging – SCoMPI – which corresponds to a unique organometallic complex showing complementary spectroscopic properties to achieve multimodal imaging in cells. As a proof of principle, we have shown that a rhenium tris-carbonyl complex with a 4-(2-pyridyl)-1,2,3-triazole (or pyta) ancillary ligand endowed with luminescent and infrared properties allowed a relevant bio-imaging correlative study using both modalities.12
In the present paper, we describe the investigation of the cellular location of an estrogen derivative conjugated with a metal-CO SCoMPI using correlative spectro-imaging. Estrogen signaling, trafficking and targeting are highly regulated mechanisms mediated by several factors. The first to be described was the α form of the nuclear estrogen receptors (ERα).27,28 In the late 1990s membrane-bound estrogen receptors were identified and among them was the 7-transmembrane G protein-coupled estrogen receptor GPR30.29 The cell lines studied here are the archetypal of breast cancer cell lines, namely MCF-7 and MDA-MB-231. They express different ER profiles since they are respectively (ER+, GPR30+) and (ER−, GPR30+).30–35 There are debates and controversies about GPR30's exact location36,37 and several authors reported its main colocalization with the endoplasmic reticulum and/or the Golgi apparatus.35,38,39
Over the past thirty years several groups have worked on the synthesis of labelled estrogens, seeking for relevant imaging of estrogen receptors.40 Nuclear ER was successfully visualized by using fluorescent tetrahydrochrysen estrogens as labels in Cos-7 cells that express elevated levels of the receptor, but not in classic ER positive breast cancer cells such as MCF-7.41 Mapping studies involved radio-labelling of estrogens with halogen isotopes (123I, 77Br, 18F),42–44 estrogen–radiometal complexes based on Tc(I) radionuclides,45–47 intrinsic fluorescence of estrogen-like molecules48 or estrogens tagged with fluorescent probes.49–55 Overall, the nuclear location was expected but rarely observed, which stresses the difficulty to visualize nuclear receptors, probably because of their low concentration. Combination of techniques in a multimodal approach would provide valuable cross-correlative information.56 Multimodal probes with complementary spectral properties, such as the SCoMPI we developed,12 are relevant candidates to achieve this goal.
3-Methoxy-17α-ethynylestradiol or mestranol (MW 310.4) (Scheme 1) is the estrogen component of some oral contraceptive formulations and a prodrug leading to its active metabolite 17α-ethynylestradiol.57,58 It was reported that bulky groups could be introduced without modifying the binding affinity at the 17α-position.59,60 It was tagged using click chemistry at position 17 by a metal-CO SCoMPI bearing an azide (MW 534.9), leading to 1 (MW 845.3). Using the bimodal properties of the SCoMPI complex, several techniques based on luminescence and vibrational spectroscopies were implemented to explore its cellular distribution in MCF-7 and MDA-MB-231 breast cancer cells through a correlative imaging approach.
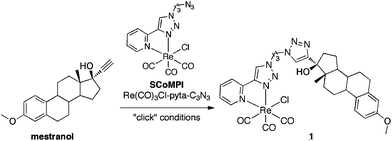 | ||
| Scheme 1 | ||
Luminescence studies were performed using wide field and confocal fluorescent microscopies as well as synchrotron radiation UV spectromicroscopy (DISCO beam line, SOLEIL, Paris). Vibrational imaging was performed using synchrotron-based radiation FTIR with a raster scanning recording (SMIS beam line, SOLEIL, Paris), synchrotron-based multiple beam FTIR using a parallel recording with a focal plane array detector (IRENI beam line, SRC Madison), confocal Raman (CBMN, Bordeaux I) and AFMIR (CLIO centre, Paris-Sud university) spectromicroscopies. These techniques were applied to track 1 in fixed MCF-7 and/or MDA-MB-231 cells. All the mappings were consistent with one another and in agreement with colocalization of 1 with the Golgi apparatus.
2 Results and discussion
2.1 Synthesis and characterization of 1
Mestranol (Scheme 1) bears a terminal alkyne in the 17α-position that can be used for Huisgen reaction and its copper-catalyzed click version (CuACC)61,62 and thus was readily coupled to the azide group from Re(CO)3Cl-pyta-C3N3. The two enantiomers of the facial Re(CO)3Cl-pyta-C3N3 (ref. 63–65) were not separated before coupling to the chiral mestranol, resulting in two diastereoisomers. These two diastereoisomers, bearing the same facial Re(CO)3Cl-pyta moiety, are expected to show similar IR and luminescence properties. Indeed, as expected for a (pyta)Re(CO)3 central core, three bands are observed in the IR-spectrum in the 1800–2200 cm−1 range and a unique large MLCT band in the UV-visible absorption spectrum. In addition, the 1H and 13C NMR signal sets are very similar for the two diastereoisomers.60 The diastereoisomeric mixture was used without any separation and will be referred to as conjugate 1 (Scheme 1).Conjugates involving metal-carbonyl complexes and estrogen derivatives are reported in the literature45,46,60,65–67 but, as far as we know, they were never used for infrared imaging. We previously showed that the rhenium tris-carbonyl moiety was reliable for sub-cellular IR-imaging5,11 using the strong infrared absorptions of CO stretching (E and A1-bands in C3v symmetry, respectively at 1915 cm−1 and 2025 cm−1).68 It should be noted that the triple bond stretching vibration in the mestranol is far weaker than that of the CO in 1 and hence inappropriate for cellular imaging (see Fig. S2†). Rhenium-pyta tris-carbonyl complexes are luminescent upon excitation in a metal-to-ligand charge transfer (MLCT) broad band from 320–350 nm that deexcites at ca. 510 nm.69–71 As previously described,12 this luminescence can be used for sub-cellular mapping. The spectral characteristics of 1 in IR and luminescence are shown on Fig. 1 (B and C frames, trace 3). It was shown to be lipophilic, with a log Po/w of 5.7 (see Section 3.1), which is higher but close to the value for mestranol alone (4.7).57 This lipophilicity value remains yet in the range of analogous compounds dedicated to estrogen receptor targeting.53,72,73
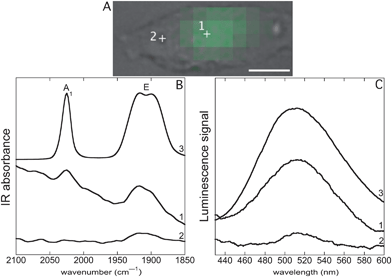 | ||
Fig. 1 SR-UV-SM and SR-FTIR-SM (Sections 3.7 and 3.8). Single MDA-MB-231 cell incubated with 1 (25 μM, 1 h), fixed and air-dried. (A) Bright field image, scale bar: 10 μm. Green pixels: intensity for the integral of the band of luminescence emission (450–550 nm) recorded by SR-UV-SM after excitation at 350 nm. (B) (1) and (2) SR-FTIR-SM spectra recorded respectively at locations 1 and 2 inside the cell, (3) FTIR spectrum of pure 1 (see Section 3.12 and Fig. S2†). (C) (1) and (2) SR-UV-SM spectra recorded respectively at locations 1 and 2 inside the cell, (3) luminescence emission after excitation at 350 nm of a solution of 1 at 10−4 M in a DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water (1:9) mixture. :water (1:9) mixture. | ||
2.2 Biological assays and effect of the temperature on cellular uptake
The in vitro effect of 1 on both hormone dependent (MCF-7) and hormone independent breast cancer cells (MDA-MB-231) was studied after 5 days of culture. On MCF-7 cells 1 shows a strong estrogenic proliferative effect at 1 μM (142% of the control) and even at the low concentration of 10 nM (157% of the control). This effect is almost identical to that with 1 nM of estradiol (174% of the control) and is consistent with the estrogenic effect of mestranol at 10 nM.74 These results indicate that despite the presence of a bulky substituent at the 17α-position of estradiol, the complex is still able to interact with the estrogen receptor. This is not really surprising, as we have previously shown that the 17α-position of estradiol is almost the only position in the steroid that is able to accommodate bulky substituents.59 In MDA-MB-231 cells 1 is toxic but in rather high concentrations (IC50 = 4.6 μM ± 1.8).The cellular uptake of the unconjugated probe Re(CO)3Cl-pyta-C3N3 (see Scheme 1) was studied by fluorescence microscopy and synchrotron radiation UV spectromicroscopy and was almost insignificant (not shown), which suggests that the mestranol moiety is involved in the uptake of 1. The uptake of 1 was also further examined at 4 °C, a temperature that abolishes active transport mechanisms75 and where only passive diffusion occurs,22,76,77 and compared with uptake at 37 °C. Quantification of the cellular uptake was performed by determining the mean perinuclear fluorescence intensity per cell after incubation with 1 at 25 μM, at 4 °C or at 37 °C (see Section 3.6). Internalization of 1 was observed at 4 °C but this represented only 11% of the uptake at 37 °C (Fig. S3-left†). These results were compared with the uptake of the compound Re(CO)3Cl-pyta-C12N3 that we previously described.12 This compound was shown to display a log Po/w of 7.3 (the same method as that used for 1). Interestingly, Re(CO)3Cl-pyta-C12N3 displays an uptake at 4 °C representing 54% of the uptake at 37 °C (Fig. S3-right†). This indicates that 1 enters cells by active transport to a larger extent than Re(CO)3Cl-pyta-C12N3 that does not bear the mestranol moiety, and suggests further that the penetration of 1 involves the mestranol moiety and its recognition.
In the following, we describe spectromicroscopy analyses of either MCF-7 or MDA-MB-231 breast cancer cell lines incubated with 1 under the same conditions (25 μM, 1 h, 37 °C) using several luminescent and IR imaging techniques. Note that most of the images were recorded with cells fixed and then air-dried, conditions required for IR-mapping. In two cases, the cells were fixed and then slides were mounted. Conditions are indicated in the figure captions and Sections 3.4 and 3.5.
2.3 Synchrotron UV and IR spectromicroscopies (SR-UV-SM and SR-FTIR-SM)
Fig. 1 shows SR-FTIR-SM and SR-UV-SM analyses of a single MDA-MB-231 cell incubated with 1. The advantage of the synchrotron-based light is the highly focused beam hence allowing to record spectra at the subcellular level. At location 1, an intense luminescence emission of 1 obtained by excitation at 350 nm was recorded (Fig. 1C, trace 1). The spectrum matched with that of the pure compound in solution (Fig. 1C, trace 3). At location 2, a very weak signal was recorded (Fig. 1C, trace 2). To generate a map of 1, the luminescent band obtained by excitation at 350 nm was integrated from 450 to 550 nm and Fig. 1A shows the location of 1 as green pixels the intensity of which corresponds to the integration values. A similar location was observed using the IR signals of 1 (E and A1-bands, respectively at 1915 and 2025 cm−1) which were stronger where the luminescence signal was the highest (Fig. 1B, traces 1 and 2).Fig. 2 shows SR-FTIR-SM and SR-UV-SM mappings of 1 obtained from a single MCF-7 cell incubated with 1. An IR-map was generated;11,12 as previously described, the absorbance of a given IR-band was integrated—see limits of integration indicated in figure captions—and colored pixels are those showing more than 80% of the maximum integration value. These colored areas will be referred to as hot spots—see also Fig. S4† for a mapping with the full range of integrated absorbance values. Showing IR hot spots superimposed on the bright field image enables a clear comparison between IR and luminescent imaging on the same cell.
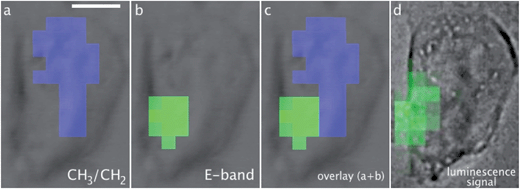 | ||
| Fig. 2 SR-FTIR-SM and SR-UV-SM (Sections 3.7 and 3.8). A single MCF-7 cell incubated with 1 (25 μM, 1 h), fixed and air-dried. Scale bar: 10 μm. Pixel size 3 × 3 μm2. Hot spots of the integral of IR bands: (a) CH3/CH2 ratio (using the CH3 asymmetric stretching band from 2986 to 2948 cm−1 and the CH2 asymmetric stretching band from 2948 to 2897 cm−1), (b) E-band (1940–1879 cm−1), (c) overlay CH3/CH2 + E-band. Integration of the band of luminescence (d) (450–550 nm) recorded by SR-UV-SM after excitation at 350 nm. Note that the bright field image in (d) was collected on a different microscope compared to those in (a–c). | ||
The IR-map using the E-band of 1 (Fig. 2b) matched with that of the luminescence signal of 1 (Fig. 2d). Interestingly, as previously discussed, the nucleus can be located without any trackers using CH3 and CH2 IR-bands;78 the ratio of the CH3 asymmetric stretching band and the CH2 asymmetric stretching band is the highest at the nucleus and the lowest in the lipid-rich organelles including the Golgi apparatus and the endoplasmic reticulum. Fig. 2a shows in blue the hot spot of the CH3/CH2 ratio, which thus corresponds to the nucleus. Fig. 2c hence indicates a perinuclear location of 1.
2.4 Confocal Raman microspectroscopy and imaging
MDA-MB-231 cells incubated with 1 were also analysed by confocal Raman microspectroscopy (see Section 3.10). The Raman spectrum of 1 in the solid state is shown in Fig. 3d (top black trace) and in Fig. S5.† The characteristic vibrational modes of the Re(CO)3 moiety were observed at 1915 cm−1 and 2032 cm−1.12 Additional bands from 1 are observed between 400 and 1800 cm−1, assigned to the stretching or the bending modes of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, NN, C–C and C–N bonds. An optical image of an MDA-MB-231 cell incubated with 1 is shown in Fig. 3a. The Raman image based on the CO stretching intensity gave the location of 1 in the same cell (Fig. 3b). Since CO-bands are weaker in Raman spectroscopy than in infrared spectroscopy it can be useful to take into account the other characteristic Raman bands of 1. The determination of the distribution of 1 was thus improved using the Hierarchical Cluster Analysis (HCA).79 A mask was selected based on the characteristic vibrational bands of 1 (Fig. S5†) and the HCA process was performed with five cluster areas in the mapping. Fig. 3c displays a color representation of the HCA calculated cluster areas. The average spectrum of each cluster area resulting from the HCA uses the same color code (Fig. 3e). The bottom black spectrum shows no significant band, as expected due to the location out of the cell; it mainly reveals characteristic bands of the glass slide where the cells grew. From cyan to green, an increase in intensity in the CO bands was observed, showing an inhomogeneous distribution of 1 inside the cell. Raman signals of 1 were weak in the blue and cyan parts of the cell, but higher in the red and green parts of the cell, indicating an accumulation of 1 in the red and green areas. It is noteworthy that in the average cluster spectra of the red and green parts, not only the CO stretching can be observed but also other characteristic bands of 1 at 1627 cm−1, 1590 cm−1, 1000 cm−1, 755 cm−1, 648 cm−1 and 500 cm−1 (see stars in Fig. 3e) can be observed. As expected, none of the bands was observed in the analysis of the control cell (Fig. S6†). This distribution is consistent with that obtained with the other imaging techniques, showing that 1 can be mapped by Raman spectroscopy.
C, NN, C–C and C–N bonds. An optical image of an MDA-MB-231 cell incubated with 1 is shown in Fig. 3a. The Raman image based on the CO stretching intensity gave the location of 1 in the same cell (Fig. 3b). Since CO-bands are weaker in Raman spectroscopy than in infrared spectroscopy it can be useful to take into account the other characteristic Raman bands of 1. The determination of the distribution of 1 was thus improved using the Hierarchical Cluster Analysis (HCA).79 A mask was selected based on the characteristic vibrational bands of 1 (Fig. S5†) and the HCA process was performed with five cluster areas in the mapping. Fig. 3c displays a color representation of the HCA calculated cluster areas. The average spectrum of each cluster area resulting from the HCA uses the same color code (Fig. 3e). The bottom black spectrum shows no significant band, as expected due to the location out of the cell; it mainly reveals characteristic bands of the glass slide where the cells grew. From cyan to green, an increase in intensity in the CO bands was observed, showing an inhomogeneous distribution of 1 inside the cell. Raman signals of 1 were weak in the blue and cyan parts of the cell, but higher in the red and green parts of the cell, indicating an accumulation of 1 in the red and green areas. It is noteworthy that in the average cluster spectra of the red and green parts, not only the CO stretching can be observed but also other characteristic bands of 1 at 1627 cm−1, 1590 cm−1, 1000 cm−1, 755 cm−1, 648 cm−1 and 500 cm−1 (see stars in Fig. 3e) can be observed. As expected, none of the bands was observed in the analysis of the control cell (Fig. S6†). This distribution is consistent with that obtained with the other imaging techniques, showing that 1 can be mapped by Raman spectroscopy.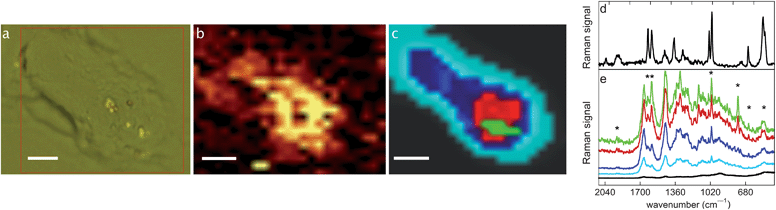 | ||
| Fig. 3 Confocal Raman microspectroscopy (Section 3.10). A single MDA-MB-231 cell incubated with 1 (25 μM, 1 h), fixed and air-dried. Scale bars: 4 μm. (a) Bright field image, the red frame corresponds to the measured area, (b) Raman image of the integration of the CO stretching band at 1926 cm−1, (c) results of the HCA analysis, (d) Raman spectrum of 1 in the solid state, and (e) cluster average spectra from HCA results (the trace color corresponds to the area in (c)). | ||
2.5 Synchrotron-based multiple beam FTIR imaging
Fig. 4 shows the FTIR average spectra of a control (trace 2) and an incubated MDA-MB-231 cell with 1 (trace 1) in the region 2150–1600 cm−1. Specific absorption bands of 1 are observed at 1915 cm−1 (E-band) and 2025 cm−1 (A1-band). The spectra demonstrate the detection of 1 inside a single MDA-MB-231 cell using an IRENI set-up. In order to determine the distribution of 1, IRENI images were developed by integration of the individual absorption bands of interest. In Fig. 5 visible images and intensity distributions of amide I, E- and A1-bands of a control cell (series A) and an incubated cell (series B) are displayed (see Section 3.11 for integration parameters). Chemical images of replicate measurements of control and incubated cells are shown in Fig. S7.† The contour colors vary between red and dark violet, where red indicates the highest and dark violet indicates the lowest value of the band integration. For better comparison of the chemical images, constant scaling of the contour colors has been applied as a visualization tool for each individual functional group for displaying the differences of the control and incubated cell. Based on the chemical images of the cell incubated with 1 (Fig. 5, series B), it can be seen that 1 was heterogeneously distributed in the cell and localized in regions of low amide absorption or partly overlaid with the amide-rich region that can be assigned to the nucleus.80 The IRENI results were thus in agreement with the previous luminescence and IR findings, pointing out a perinuclear distribution of 1.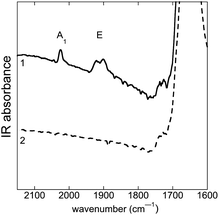 | ||
| Fig. 4 FTIR average spectra recorded with IRENI (Section 3.11) on single MDA-MB-231 cells, fixed and air-dried. (1) Single cell incubated with 1 (25 μM, 1 h); (2) single control cell. Amide I and CO ester bands appear respectively at 1650 and 1735 cm−1. | ||
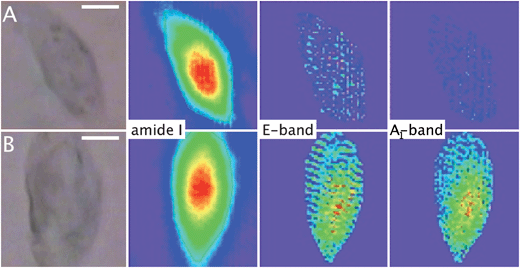 | ||
| Fig. 5 Synchrotron-based multiple beam FTIR imaging (Section 3.11). Visible and FTIR images based on the integration of specific absorption bands. Scale bars: 10 μm. (A) A single MDA-MB-231 control cell. (B) A single MDA-MB-231 cell incubated with 1 (25 μM, 1 h). See Section 3.11 for integration parameters. Constant scaling of the contour colors has been applied for each band, with red/purple equivalent to high/low absorption intensity. All cells were fixed and air-dried. | ||
2.6 Correlation between AFMIR and fluorescence imaging
MCF-7 cells grown on thin CaF2 slides and incubated with 1 were mapped using both AFMIR (see Section 3.9) and fluorescence microscopy on the same cells. The AFMIR signal upon irradiation in the E-band of 1 is shown in Fig. 6b. | ||
| Fig. 6 MCF-7 breast cancer cell incubated with 1 (25 μM, 1 h), fixed and air-dried. Scale bar: 10 μm. AFMIR imaging (Section 3.9): (a) topography of the cell recorded with the AFM (maximum of altitude 2.2 μm; cell outline drawn in gray), (b) AFMIR mapping of the E-band of 1 (laser tuned at 1920 cm−1); fluorescence imaging (Section 3.6): (c) bright field image merged with nucleus staining (DAPI, blue) and luminescence signal of 1 (green). | ||
This mapping does not correspond to all the topographic bulges recorded by the AFM tip (Fig. 6a). Interestingly the AFMIR signal for 1 matches with the luminescence signal of 1 (Fig. 6c, green). The nucleus was stained with DAPI (Fig. 6c, blue) and the mapping clearly shows that 1 has a perinuclear location.
Overall all these imaging techniques point out a perinuclear localization of 1 both in MDA-MB-231 and MCF-7 cell lines. To better characterize this location, we performed colocalization experiments using fluorescence microscopy.
2.7 Colocalization studies
Fig. 7 shows the colocalization experiments between 1 (green), a Golgi tracker (BODIPY-TR C5-ceramide, red) and DAPI (blue) in MDA-MB-231 and MCF-7 cell lines. Comparison of Fig. 7-A1 with A2 and B1 with B2 clearly showed an accumulation of 1 in a region outside the nucleus in both cell lines. This observation was confirmed by confocal microscopy (Fig. 8). It was also in agreement with the exclusion of the region with high CH3/CH2 ratio obtained in IR studies (Fig. 2c). Colocalization with the Golgi tracker was clearly shown in Fig. 7A2, A3 and B2, B3, with very high quantitative coefficients for both cell lines (Table 1).81,82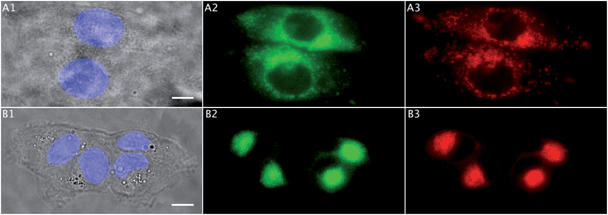 | ||
| Fig. 7 Colocalization studies (Sections 3.5 and 3.6). (A) Two MCF-7 breast cancer cells incubated with 1 (25 μM, 1 h): (A1) bright field image (scale bar: 10 μm) and the nucleus stained with DAPI (blue), (A2) 1 in green, (A3) Golgi tracker in red. (B) Four MDA-MB-231 breast cancer cells incubated with 1 (25 μM, 1 h): (B1) bright field image (scale bar: 10 μm) and the nucleus stained with DAPI (blue), (B2) 1 in green, (B3) Golgi tracker in red. All cells were fixed and slides were mounted. | ||
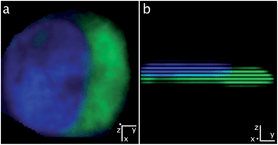 | ||
| Fig. 8 Confocal fluorescence microscopy (Section 3.6). A single MDA-MB-231 cell incubated with 1 (25 μM, 1 h). All cells were fixed and slides were mounted. Confocal images of 1 (green) and DAPI (blue): (a) along the z-axis and (b) along the x-axis (clockwise rotation of 90° along the y-axis from (a) to (b)). | ||
| Pearson's coefficient (−1 < x < 1) | Overlap coefficient (0 < x < 1) | Intensity correlation quotient (0 < x < 0.5) | |
|---|---|---|---|
| A2 and A3 | 0.90 | 0.95 | 0.35 |
| B2 and B3 | 0.95 | 0.94 | 0.41 |
The observation of a strong proliferative effect induced in MCF-7 cells with a low concentration of 1 (see Section 3.2) demonstrated that 1 was recognized by nuclear estrogen receptors, known to be responsible for this proliferative effect.83 However, the amount of nuclear estrogen receptors is known to be very low, actually in the range of a picomole per mg of DNA,84 which can explain why 1 was not detected in the nucleus with the techniques used here, due to limits in the detection sensitivity, as previously pointed out by Rickert et al.52
Images obtained in Fig. 7 were very similar to those from the literature35 for the localization of GPR30 in MCF-7 and MDA-MB-231 cells showing a perinuclear distribution. Several groups reported that GPR30 is mainly found in the internal membranes85 with a predominant expression in the endoplasmic reticulum86 or the Golgi apparatus.38,39 The purpose here was not to solve this controversy and at that point, we cannot exclude a non-specific localisation of the labelled mestranol 1. However, this multimodal approach allowed us to obtain information from independent imaging techniques about the intracellular main location of 1. The convergent results obtained are clearly in favour of accumulation of 1 in the Golgi apparatus in both cell lines.
3 Experimental section
3.1 Synthesis and characterization
Sodium azide (5 g, 7.7 × 10−2 mol) and 3-bromopropan-1-ol (Br(CH2)3OH, 5 g, 3.6 × 10−2 mol) were dissolved in water (50 mL) and heated at 60 °C overnight. After cooling to r.t., 3-azidopropan-1-ol was extracted with CH2Cl2 (3 × 100 mL). The organic fractions were combined and dried over anhydrous sulfate magnesium and evaporated to dryness. The yield was quantitative.3-Azidopropan-1-ol was directly added (1 g, 9.7 × 10−3 mol) with 2-ethynylpyridine (1 g, 9.7 × 10−3 mol) to a mixture of CH2Cl2 (5 mL) and water (5 mL) at r.t. Sulfate copper(II) pentahydrate (250 mg, 1.0 × 10−3 mol) and sodium ascorbate (500 mg, 2.0 × 10−3 mol) were then added. After overnight stirring, water (10 mL) and CH2Cl2 (10 mL) were poured into the mixture. The product was extracted with CH2Cl2 (2 × 30 mL), the organic fractions combined, dried over anhydrous sulfate magnesium and evaporated to dryness. Silica gel column chromatography with CH2Cl2:MeOH (from 1:0 to 95:5) gave 4-(2-pyridyl)-1,2,3-triazole-(CH2)3-OH (1.6 g, 7.8 × 10−3 mol, 80% yield) as a white solid. In the following, this product is referred to as pyta-(CH2)3-OH.
Pyta-(CH2)3-OH (1 g, 5 × 10−3 mol) and 4-toluenesulfonyl chloride (950 mg, 5 × 10−3 mol) were dissolved in CH2Cl2. The solution was cooled with an ice bath and potassium hydroxide (1.12 g, 2 × 10−2 mol) was poured into the mixture that remained at 0 °C for 3 h under stirring. Ice (30 g) was added and the tosylated compound was extracted with CH2Cl2 (2 × 100 mL), the organic fractions combined, dried over anhydrous sulfate sodium and evaporated to dryness. Silica gel column chromatography with CH2Cl2:MeOH (99:1) gave pyta-(CH2)3-OTs (1.6 g, 4.5 × 10−3 mol, 90% yield) as a white solid.
Pyta-(CH2)3-OTs (2.9 g, 8.1 × 10−3 mol) and sodium azide (1.65 g, 2.5 × 10−2 mol) were dissolved in DMF (50 mL) and heated at 70 °C overnight. After cooling to r.t., pyta-(CH2)3-N3 was extracted with Et2O (2 × 30 mL). Organic fractions were combined, dried over anhydrous sulfate magnesium and evaporated to dryness. The yield was quantitative. See the ESI† for the atom numbering.
Pyta-(CH2)3-N31H NMR (CDCl3, 250 MHz) δ (ppm): 8.47 (1H, d, J = 4.5 Hz, 6′-pyH), 8.10 (1H, s, 5′′-triazH), 8.06 (1H, d, J = 8.0 Hz, 3′-pyH), 7.67 (1H, m, 4′-pyH), 7.12 (1H, m, 5′-pyH), 4.42 (2H, t, J = 6.4 Hz, CH2), 3.29 (2H, t, J = 6.7 Hz, CH2), 2.11 (2H, m, CH2).
Pyta-(CH2)3-N313C NMR (CDCl3, 62.5 MHz) δ (ppm): 150.10, 149.37, 148.46, 136.99, 122.95, 122.35, 120.23, 47.99, 47.33, 29.48.
Rhenium pentacarbonyl chloride (500 mg, 1.4 × 10−3 mol) and pyta-(CH2)3-N3 (360 mg, 1.6 × 10−3 mol) were dissolved in toluene (20 mL) and heated for 3 h at 70 °C. A precipitate was formed when cooled to r.t. Filtration led to a yellow solid of Re(CO)3Cl-pyta-(CH2)3-N3 (178 mg, 3.4 × 10−4 mol, 25% yield). The conjugation with the tricarbonyl-chloro-rhenium core through the pyta ligand provided a stereogenic metal centre.64 Re(CO)3Cl-pyta-(CH2)3-N3 was obtained as a racemic mixture. See the ESI† for the atom numbering.
Re(CO)3Cl-pyta-(CH2)3-N31H NMR (CDCl3, 250 MHz) δ (ppm): 9.02 (1H, d, J = 5.4 Hz, 6′-pyH), 8.41 (1H, s, 5′′-triazH), 8.00 (1H, m, 4′-pyH), 7.87 (1H, d, J = 7.8 Hz, 3′-pyH), 7.48 (1H, m, 5′-pyH), 4.57 (2H, t, J = 6.9 Hz, CH2), 3.47 (2H, t, J = 6.1 Hz, CH2), 2.25 (2H, m, CH2).
Re(CO)3Cl-pyta-(CH2)3-N313C NMR (acetone, 75 MHz) δ (ppm): 153.07, 149.49, 148.77, 140.16, 126.13, 125.31, 122.46, 49.46, 48.01, 29.29.
Re(CO)3Cl-pyta-(CH2)3-N3 (178 mg, 3.4 × 10−4 mol) and 3-methoxy-17α-ethynylestradiol (or mestranol) (100 mg, 3.2 × 10−4 mol) were dissolved in a mixture of CH2Cl2 (5 mL) and water (5 mL) at r.t. Sulfate copper(II) pentahydrate (8 mg, 3.2 × 10−5 mol) and sodium ascorbate (13 mg, 6.5 × 10−5 mol) were then added. After overnight stirring at r.t., water (10 mL) and CH2Cl2 (10 mL) were poured into the mixture. The product was extracted with CH2Cl2 (2 × 30 mL), the organic fractions combined, dried over anhydrous sulfate sodium, and evaporated to dryness. Silica gel column chromatography with AcOEt:acetone (from 1:0 to 7:3) gave 1 (180 mg, 2.1 × 10−4 mol, 66% yield) as a pale yellow solid.
Because of the facial coordination geometry of the Re complex and the chirality of the steroid substituent, a mixture of diastereoisomers was formed, in a 1:1 ratio. Attempts to separate these two isomers by chromatography were unsuccessful, as reported in the literature for a similar derivative.63
HPLC purity was checked with a Dionex C18 analytical column. Using a CH3CN:H2O (0.1% TFA) mixture from 2:3 to 1:0 in 30 minutes, retention times of the two diastereoisomers of 1 were 14.89 and 15.01 minutes (7:3 for CH3CN:H2O). They were not isolated separately and the integration of both peaks is >95% of the whole chromatogram (see Fig. S1†).
We report below 1H and 13C NMR signals for the diastereoisomeric mixture. The integrations for 1H NMR signals are given for the two diastereoisomers. The sign “*” indicates protons with different chemical shifts in the two diastereoisomers and the indices a and b refer to the two diastereoisomers. The major shifts are noticed for 1H present on carbons 5′′ (ca. 8.6 ppm) and 13′′ (ca. 7.6 ppm) from the triazol rings. See the ESI† for the atom numbering.
11H NMR (CDCl3, 300 MHz) δ (ppm): 8.95–8.93 (2H, da (J = 4.2 Hz) and db (J = 4.5 Hz), 6′-pyH*), 8.69 and 8.64 (2H, sa and sb, 5′′-triazH*), 7.99–7.92 (4H, m, 4′ and 3′-pyH*), 7.62 and 7.60 (2H, sa and sb, 13′′-triazH*), 7.43–7.39 (2H, m, 5′-pyH*), 7.08 (2H, d, 1-ArH, J = 8.6 Hz), 6.63 (2H, dd, 2-ArH, J = 8.6 Hz, J = 2.7 Hz), 6.60 (2H, d, 4-ArH, J = 2.7 Hz), 4.53 (4H, m, -(CH2)-triaz), 4.42 (4H, m, -(CH2)-triaz), 3.75 (6H, s, 19-CH3), 2.82 (4H, m, 6-(CH2)), 2.60 (4H, m, 7′′-(CH2)), 2.35 (2H, m), 2.13 (4H, m, -CH2-, -CH-), 1.94 (6H, -CH2-, -CH-), 1.65–1.30 (12H, m, -CH2-, -CH-), 1.03 (6H, s, 18-(CH3)), 0.68 (2H, m, -CH2-).
113C NMR (CDCl3, 75 MHz) δ (ppm): 197.17 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O), 196.03 (CO), 188.82 (CO), 157.55 (3-C), 154.22 (12′′-C), 153.04 (6′-pyC), 149.14 (2′-pyC), 148.87 (4′′-triazC), 139.64 (4′-pyC), 137.95 (5-C), 132.58 (10-C), 126.15 (1-C), 125.99 (5′-pyC), 123.84 (5′′-triazC), 122.60 (13′′-triazC), 122.49 (3′-pyC), 113.91 (4-C), 111.52 (2-C), 82.34 (17-C), 55.21 (19-C), 49.20 (-CH2-triaz), 48.70 (14-C), 47.46 (13-C), 46.60 (-CH2-triaz), 43.43 (9-C), 39.54 (8-C), 38.12 (12-C), 33.14 (16-C), 29.99 (6-C), 29.77 (7′′-C), 27.39 (11-C), 26.30 (7-C), 23.46 (15-C), 14.30 (18-C).
O), 196.03 (CO), 188.82 (CO), 157.55 (3-C), 154.22 (12′′-C), 153.04 (6′-pyC), 149.14 (2′-pyC), 148.87 (4′′-triazC), 139.64 (4′-pyC), 137.95 (5-C), 132.58 (10-C), 126.15 (1-C), 125.99 (5′-pyC), 123.84 (5′′-triazC), 122.60 (13′′-triazC), 122.49 (3′-pyC), 113.91 (4-C), 111.52 (2-C), 82.34 (17-C), 55.21 (19-C), 49.20 (-CH2-triaz), 48.70 (14-C), 47.46 (13-C), 46.60 (-CH2-triaz), 43.43 (9-C), 39.54 (8-C), 38.12 (12-C), 33.14 (16-C), 29.99 (6-C), 29.77 (7′′-C), 27.39 (11-C), 26.30 (7-C), 23.46 (15-C), 14.30 (18-C).
HRMS+ (ESI): calcd for ReC34H37O5N7ClNa ([M + Na]+): m/z 868.1987. Found: m/z 868.2021.
Log Po/wof1 was estimated by HPLC chromatography following the recommendations of Minick87 and Pomper88 using a C-18 silica column (Kromasyl, Interchrom) and uracil as an internal reference. Organic and aqueous phases were used in several proportions (n = 5). The organic portion of the mobile phase was composed of methanol containing 0.25% (v/v) 1-octanol. The aqueous portion was prepared from octanol-saturated water containing 0.02 M MOPS buffer, and 0.15% (v/v) n-decylamine, and adjusted to pH 7.4. We found: log Po/w (1) = 5.7 and log Po/w (Re(CO)3Cl-pyta-C12N312) = 7.3.
3.2 Biological assays
MDA-MB-231 and MCF-7 breast cancer cell lines obtained from the Human Tumour Cell Bank were used for the experiments. Cells were maintained in monolayer culture in DMEM with phenol red/Glutamax I, supplemented with 9% of decomplemented fetal calf serum and 0.9% kanamycin, at 37 °C in a 5% CO2 air humidified incubator. For proliferation assays, cells were seeded in 24-well sterile plates with 1.5 × 104 cells of MDA-MB-231 and 3 × 104 cells of MCF-7 in 1 mL of DMEM without phenol red, supplemented with 9% of fetal calf serum desteroided on dextran charcoal, 0.9% Glutamax I and 0.9% kanamycin, and grown for 24 h. The following day (D0), 1 mL of the same medium containing 1 dissolved in DMSO was added to the plates (final volumes of DMSO: 0.1%; 4 wells for each condition). After three days (D3), the incubation medium was removed and 2 mL of fresh medium containing 1 was added. At D3, D4, D5 and D6, the protein content of each well was quantified by methylene blue staining as follows. Cell monolayers were fixed and stained for 1 h in methanol with methylene blue (2 mg mL−1), and then washed thoroughly with water. Two milliliters of HCl (0.1 M) was then added, and the plate was incubated for 1 h at 37 °C. Then the absorbance of each well was measured at 655 nm with a Biorad microplate reader. The results are expressed as the percentage of proteins versus the control. The estrogenic activity (on MCF-7) and cytotoxicity (IC50 on MDA-MB-231) were both determined at D5. Experiments were performed at least in duplicate.3.3 Nature of the slides
Glass slides were used for wide field and confocal fluorescence imaging and Raman microspectroscopy. Calcium fluoride (CaF2) slides (purchased from Crystran) were used for synchrotron radiation FTIR (SMIS and IRENI, 1 and 0.2 mm thick) and UV spectromicroscopies (DISCO, 0.2 mm thick). These 0.2 mm thin CaF2 slides are useful windows for multimodal imaging as they are transparent from 190 nm to 10 μm; it is then possible to study the same cell by both UV-visible and IR techniques (see Fig. 1, 2 and 6).3.4 Cell culture for imaging
Both control and treated cells were processed in a similar way. They were seeded on slides (deposited in 35 × 10 mm Petri dishes) in order to reach confluency after 48 h of incubation at 37 °C under an atmosphere of 95% air/5% CO2. The nature of the slides for each imaging technique is described in Section 3.3. The medium was removed and fresh growth medium (2 mL DMEM without phenol red and supplemented as in Section 3.2) was added to each flask of control cells. In the case of treated cells, 2 mL of a solution of 1 in fresh growth medium (25 μM prepared from a 5 × 10−3 M stock solution in DMSO) were added. The cells were incubated at 37 °C under an atmosphere of 95% air/5% CO2 for 1 h. The medium was then removed and the cells were washed twice with phosphate buffered saline (D-PBS, 1×, 2 mL). The cells were fixed with 4% paraformaldehyde (1.5 mL) for 8 minutes at room temperature and washed once with D-PBS (1×, 2 mL) and once with pure water (2 mL). Slides were air-dried for IR-imaging and mounted using the Vectashield solution (H-1000, Vector Laboratories) just after the last water washing for luminescence imaging.3.5 Fluorescent organelle staining
For colocalization of the Golgi tracker (BODIPY TR C5-ceramide) and 1, the experiment was performed as follows: growth medium (DMEM without phenol red and supplemented as in Section 3.2) was removed and cells were incubated at 4 °C with BODIPY TR C5-ceramide complexed to BSA (B-34400, Invitrogen) (2.7 μM in growth medium for 30 minutes). The medium was then removed and the cells were washed twice with cold phosphate buffered saline (D-PBS, 1×, 2 mL). 2 mL of a 25 μM solution of 1 in fresh growth medium (prepared from a 5 × 10−3 M stock solution in DMSO) were then added. The cells were incubated at 37 °C under an atmosphere of 95% air/5% CO2 for a period of 1 h. The medium was then removed and the cells were washed twice with phosphate buffered saline (D-PBS, 1×, 2 mL). Cells were fixed with 4% paraformaldehyde (1.5 mL) for 8 minutes at room temperature and washed once with D-PBS (1×, 2 mL) and once with pure water (2 mL). Slides were mounted using the Vectashield solution (H-1000, Vector Laboratories). The nuclei were stained with DAPI (Invitrogen, 15 nM in water) directly on fixed cells, when slides were on the microscope motorized stage.3.6 Fluorescence imaging and study of the effect of temperature on cellular uptake
In-cell fluorescence imaging of 1 was performed using an IX71 (Olympus) microscope equipped with a CCD (Orca-ER, Hamamatsu, Corporation, Sewickley, PA) and X60 (Plan Apo, NA 1.42) objective. 1 was detected using an appropriate filter set (excitation D350/50x; beam splitter 400DCLP; emission HQ560/80m; Chroma Technology) and excited using a Hg lamp (100 W) attenuated by a neutral density filter (ND-1). The DAPI-stained nucleus was located using a DAPI filter set (excitation D350/50x; beam splitter 400DCLP; emission D460/50m; Chroma Technology) and the BODIPY-stained Golgi using a Texas Red filter set (excitation ET560/40x; beam splitter T585LPXR; emission ET630/75m; Chroma Technology). Microscope settings and functions were controlled using Simple PCI software (Hamamatsu). Image analysis was performed using ImageJ Software and Simple PCI software. Colocalization coefficients81,82 were calculated using ImageJ plugin Colocalization Indices. Confocal fluorescence images were taken on a Zeiss LSM 710 confocal microscope using an X63 (NA 1.40) objective and 405 nm laser excitation. 3D reconstruction of the z-stacks was made with ImageJ software.To study the effect of temperature on the cellular uptake of 1, MDA-MB-231 cells were incubated for 1 h either at 37 °C (T1) or at 4 °C (T2) with a solution of 1 at 25 μM (solution previously cooled to 4 °C for the T2 experiment). The medium was then removed and the cells washed twice with D-PBS (1×, 2 mL). In the case of T2, this step was performed at 4 °C with cold D-PBS. Cells were then fixed with 4% paraformaldehyde (1.5 mL) for 8 minutes at room temperature and washed once with D-PBS (1×, 2 mL) and once with pure water (2 mL). Slides were mounted using the Vectashield solution (H-1000, Vector Laboratories) just after the last water washing for luminescence imaging. Two independent experiments were performed, each one in duplicate. A series of snapshots using the adequate filter set to monitor 1 (see above) was recorded. For each image, the mean perinuclear luminescence intensity per cell corrected for the background was determined (using ImageJ software). For the three conditions (control/incubation of 1 at 4 °C/incubation of 1 at 37 °C), mean values and error bars were calculated over a large number of cells, respectively 85, 263, 289 (Fig. S3-left†). For the compound Re(CO)3Cl-pyta-C12N3 that we previously described,12 the same protocol was used, using the same filter set as its luminescent properties are identical to 1. For the three conditions (control/incubation at 4 °C/incubation at 37 °C), mean values and error bars were calculated over a large number of cells, respectively 85, 248, 247 (Fig. S3-right†).
3.7 Synchrotron radiation FTIR spectromicroscopy (SR-FTIR-SM)
Spectra were recorded at the SMIS beamline (Synchrotron SOLEIL, Saint Aubin, France) which exploits the edge and bending radiation of a bending magnet.89 Spectra were recorded in transmission mode on a Nicolet Continuum XL microscope (Thermo Fisher) equipped with a 50 × 50 μm2 liquid nitrogen cooled MCT/A detector, a 32X/NA 0.65 Schwarzschild objective, a Prior XYZ motorised stage, and coupled to a Nicolet 5700 FTIR spectrometer (Thermo Fisher) equipped with a Michelson interferometer and a KBr beam splitter. The confocal aperture was set at 6 × 6 μm2 and the microscope was operated in semi-confocal mode. Spectra were recorded with 200–250 scans at 6 cm−1 resolution in the 1040–3700 cm−1 range. Maps were recorded by raster scanning the cells with steps of 3 μm in X and in Y with 180–250 scans at 6 cm−1 resolution in each square. Chemical maps were created with the Omnic software (Thermo Fisher) by measuring and plotting the integral of specific bands (in Fig. 2, the following integration limits were used, E: 1940–1879 cm−1, amide I: 1724–1584 cm−1, CH3 asym.: 2986–2948 cm−1 and CH2 asym.: 2948–2897 cm−1 for the CH3/CH2 ratio). Pixels showing more than 80% of the maximum value for an IR-band are referred to as hot spots and are shown in Fig. 2. The complete mappings with the full range of integration values are presented in Fig. S4.†3.8 Synchrotron radiation UV spectromicroscopy (SR-UV-SM)
Spectra were recorded at the DISCO beamline (Synchrotron SOLEIL, Saint Aubin, France). CaF2 thin slides (see Section 3.3) with fixed cells were deposited onto an inverted glycerin immersion objective (X40 Ultrafluar, CARL ZEISS). Excitation was provided by the fully tuneable excitation from a bending magnet at Synchrotron SOLEIL90 (350 nm excitation with a 0.2 nm bandpass). Rastering of the sample allows us to record (x,y,λ,I) maps with 3 × 3 μm2 pixel size, with 100–120 s acquisition time per spectrum. The fluorescence emission spectrum of the selected pixels was recorded by a spectrograph.913.9 AFMIR
AFMIR is an emerging and cutting-edge near-field technique92 using an AFM (Atomic Force Microscope) and a tuneable infrared pulsed laser to record spatially resolved absorption measurements.93–95 The AFM tip is in contact with the sample that is illuminated by a pulsed laser beam through a ZnSe prism. The laser wavelength is tuned to an IR-absorption band of the molecule of interest and when the laser pulse occurs, the temperature increases with local temporary deformations. The AFM-tip detects these local deformations and starts to oscillate. The maximal amplitude of this oscillation, which corresponds to the AFMIR signal, is proportional to the local absorbance.96 This technique has a resolution from 100 nm to 20 nm (ref. 97) thus allowing sub-cellular IR-mapping of biological samples.98 Here we used thin CaF2 slides (see Section 3.3) laid on the upper surface of the ZnSe prism with paraffin oil in between to make a good optical contact and avoid evanescent waves in the air gap. The AFM was a FlexAFM from the Nanosurf company, and the tuneable laser source was the free electron laser CLIO (French Free Electron Laser facility based in the Laboratoire de Chimie Physique at the Université Paris-Sud at Orsay).3.10 Confocal Raman microspectroscopy and imaging
Raman spectra were recorded using a WITec (Ulm, Germany) Alpha300RS confocal Raman microscope. The excitation wavelength of 532 nm was provided by a frequency-doubled Nd:YAG laser. The beam was focused on the sample using an Olympus (100×/0.95 NA) objective, and the power at the sample was close to 1 mW. The sample was located on a piezoelectrically driven microscope scanning stage, with a x, y resolution of 5 nm, and a z-resolution of 2 nm. The integration time for each spectrum was 2 s. A spectrum was recorded every 1 μm along the x and y directions. The image resulted in about 625 (25 × 25) spectra and 1250 s total acquisition time.3.11 Synchrotron-based multiple beam FTIR imaging
The synchrotron-based multiple beam FTIR imaging instrument called IRENI, as recently implemented at the Synchrotron Radiation Center (Stoughton, Wisconsin, USA), couples a synchrotron source extracting an entire bending magnet of radiation to a FTIR microscope (Bruker Hyperion 3000) equipped with a FPA detector (128 × 128 pixels).99,100 The imaging system extracts 12 beams from a bending magnet. A set of mirrors creates a bundle of a 3 × 4 beam matrix that is able to illuminate 96 × 96 pixels of a FPA detector, where each pixel corresponds to 0.54 × 0.54 μm2 area of the sample. Thus, several thousands of individual FTIR spectra are collected concurrently for a 52 × 52 μm2 sample area within a few minutes allowing rapid, high spatially and spectrally resolved chemical imaging and thereby providing chemical images of the investigated sample area. The lateral spatial resolution achievable with this widefield imaging approach is equivalent to or is better than the confocal geometry employed at traditional synchrotron beamlines when the effective sample area evaluated with defining apertures is larger than 1 μm2.101 For the present investigations, FTIR data were collected in transmission mode as hyperspectral cubes (x, y, Abs(n)) using a 74× objective (NA = 0.65) and a 15× condenser (NA = 0.65). The size of the FPA detector was 64 × 64 pixels and simultaneously 4096 single FTIR spectra were recorded corresponding to 34 × 34 μm2 area with a pixel resolution of 0.54 μm. The FTIR data were obtained over the range of 3800–1000 cm−1 and were generated from co-adding 128 scans at 4 cm−1 spectral resolution. The following integration parameters were used in Fig. 5 and S7† (with straight baselines from the integration endpoints): amide I: 1680–1612 cm−1, E: 1932–1890 cm−1 and A1: 2033–2013 cm−1. The resulting spectra and FTIR imaging data were evaluated by using homemade software IRidys (https://www.IRidys.com, accessed December 2012 4th) running under IGOR Pro 6.22.3.12 FTIR spectra acquisition
Spectra were recorded on a Perkin Elmer Spectrum 100 equipped with an ATR sampling accessory and analysed using Perkin Elmer Spectrum software. The FTIR spectra of a thin film of 1 obtained after evaporation of a solution of 1 in methanol and of mestranol as a solid are shown in Fig. S2.†3.13 UV-visible absorption and luminescence emission in vitro
Solutions of 1 in different organic solvents were studied in quartz cuvettes with a Varian Cary 300 Bio UV-Visible Spectrophotometer. Spectral analysis was performed using Cary Win UV software. A Jasco FP-8300 Spectrofluorimeter (with Spectra Manager software) was used to monitor emission properties of 1.4 Conclusion
In the intricate context of the estrogen receptor location, we report here the successful multimodal imaging of mestranol coupled with a single rhenium-pyta carbonyl bimodal core using a large set of spectroscopic techniques. Luminescent and vibrational properties of the SCoMPI enabled the detection of 1 inside fixed MCF-7 and MDA-MB-231 breast cancer cells. 1 displays estrogenic activity, suggesting the targeting of nuclear estrogen receptors. At that point, the presence of 1 at low concentrations in the nucleus cannot be excluded, but there is a clear accumulation in a perinuclear location evidenced by several independent imaging techniques. Further colocalization studies using a fluorescent Golgi tracker clearly showed colocalization of 1 with the Golgi apparatus in both cell lines. However non-specific accumulation in the Golgi that could play the role of a reservoir cannot be excluded, especially when the nuclear estrogen receptors are found to be in low concentrations. What is significant is that the images obtained from these independent techniques consistently depict accumulation inside the Golgi apparatus.These results clearly demonstrate that SCoMPIs are relevant low-molecular weight and easy-to-conjugate probes, enabling the gathering of reliable information from independent imaging techniques. By exploiting the different capabilities of the various techniques i.e. high lateral resolution in fluorescence or micro-Raman imaging, photo-bleaching-free synchrotron-based micro-FTIR using raster scanning or focal plane array detection, and high resolution in AFMIR, this LRe(CO)3 probe is a valuable tool for answering biology-related questions concerning locations with versatile constraints.
This multiple capability of a single probe is an asset as several questions can be addressed by its unique conjugation with a biomolecule.
Abbreviations
| AFMIR | Atomic Force Microscope-InfraRed |
| BSA | Bovine Serum Albumin |
| BODIPY | boron-dipyrromethene (dye) |
| CLIO | Centre Laser Infra-Rouge d’Orsay |
| DAPI | 4’,6-diamidino-2-phenylindole (nucleus tracker) |
| DISCO | Dichroism, Imaging and mass Spectrometry for Chemical and biOlogical systems |
| DMEM | Dulbecco Modified Eagle Medium |
| DMSO | DiMethylSulfOxide |
| DPBS | Dulbecco’s Phosphate Buffer Saline |
| ER | Nuclear Estrogen Receptor |
| GPR30 | G-protein coupled estrogen receptor |
| HCA | Hierarchical Cluster Analysis |
| HPLC | High Performance Liquid Chromatography |
| HRMS+ (ESI) | High Resolution Mass Spectrometry, positive mode, ElectroSpray Ionisation |
| IRENI | InfraRed ENvironmental Imaging |
| MCF-7 and MDA-MB-231 | Breast cancer cell lines |
| MLCT | Metal-to-Ligand Charge Transfer |
| MOPS | 3-(N-morpholino)propanesulfonic acid |
| MW | Molecular Weight |
| Pyta | 4-(2-pyridyl)-1,2,3-triazole |
| SCoMPI | Single Core Multimodal Probe for Imaging |
| SMIS | Spectroscopy and Microscopy in the Infrared using Synchrotron |
| SR-FTIR-SM | Synchrotron Radiation Fourier Transform InfraRed SpectroMicroscopy |
| SR-UV-SM | Synchrotron Radiation UV SpectroMicroscopy; synchrotron beamlines |
Acknowledgements
This work is based on research conducted at the Ecole Normale Supérieure (ENS), CNRS (UMR7203, 8640, 8000, 7223 and 5248), Université Pierre et Marie Curie Paris 6, Université Paris-Sud and Université de Bordeaux. These institutions are gratefully acknowledged for their financial support. In addition, ENS is acknowledged for SC's PhD fellowship, the Groupe Français de Chimie Bio-inorganique is acknowledged for SC's international fellowship for experiments at SRC, and CNRS is acknowledged for financial support in the context of programme PCV prise de risque. We thank the SOLEIL committee for beamtime and financial support in the conduct of experiments at SOLEIL (project 20111026) and the CLIO committee for beamtime for AFMIR experiments. This work is based on research conducted at the IRENI beamline the construction and development of which was supported by the U.S. National Science Foundation under award MRI-0619759. This work was supported by the U.S. National Science Foundation under grant CHE-1112433. The Synchrotron Radiation Center is primarily funded by the University of Wisconsin-Madison with supplemental support from facility Users and the University of Wisconsin-Milwaukee.Notes and references
- S. G. Kazarian and K. L. A. Chan, Analyst, 2013, 138, 1940–1951 RSC.
- L. M. Miller and P. Dumas, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 846–857 CrossRef CAS.
- H. Amrania, A. P. McCrow, M. R. Matthews, S. G. Kazarian, M. K. Kuimova and C. C. Phillips, Chem. Sci., 2011, 2, 107–111 RSC.
- M. K. Kuimova, K. L. A. Chan and S. G. Kazarian, Appl. Spectrosc., 2009, 63, 164–171 CrossRef CAS.
- C. Policar, J. B. Waern, M. A. Plamont, S. Clède, C. Mayet, R. Prazeres, J.-M. Ortega, A. Vessières and A. Dazzi, Angew. Chem., Int. Ed., 2011, 50, 860–864 CrossRef CAS.
- A. Vessières, S. Top, A. A. Ismail, I. S. Butler, M. Louer and G. Jaouen, Biochemistry, 1988, 27, 6659–6665 CrossRef.
- G. R. Stephenson, in Bioorganometallics: Biomolecules, Labeling, Medicine, ed. G. Jaouen, Wiley-VCH, Weinheim, 2006, pp. 215–262 Search PubMed.
- M. Salmain, A. Vessières, A. Varenne, P. Brossier and G. Jaouen, J. Organomet. Chem., 1999, 589, 92–97 CrossRef CAS.
- A. Marcelli, A. Cricenti, W. M. Kwiatek and C. Petibois, Biotechnol. Adv., 2012, 30, 1390–1404 CrossRef CAS.
- K. V. Kong, W. Chew, L. H. K. Lim, W. Y. Fan and W. K. Leong, Bioconjugate Chem., 2007, 18, 1370–1374 CrossRef CAS.
- S. Clède, F. Lambert, C. Sandt, Z. Gueroui, N. Delsuc, P. Dumas, A. Vessières and C. Policar, Biotechnol. Adv., 2013, 31, 393–395 CrossRef.
- S. Clède, F. Lambert, C. Sandt, Z. Gueroui, M. Refregiers, M.-A. Plamont, P. Dumas, A. Vessieres and C. Policar, Chem. Commun., 2012, 48, 7729–7731 RSC.
- I. S. Butler, R. P. Kengne-Momo, G. Jaouen, C. Policar and A. Vessieres, Appl. Spectrosc. Rev., 2012, 47, 531–549 CrossRef CAS.
- K. Meister, J. Niesel, U. Schatzschneider, N. Metzler-Nolte, D. A. Schmidt and M. Havenith, Angew. Chem., Int. Ed., 2010, 49, 3310–3312 CrossRef CAS.
- P. Hildebrandt, Angew. Chem., Int. Ed., 2010, 49, 4540–4541 CrossRef CAS.
- M. Patra and G. Gasser, ChemBioChem, 2012, 13, 1232–1252 CrossRef CAS.
- M. Wrighton and D. L. Morse, J. Am. Chem. Soc., 1974, 96, 998–1003 CrossRef CAS.
- L. Sacksteder, A. P. Zipp, E. A. Brown, J. Streich, J. N. Demas and B. A. DeGraff, Inorg. Chem., 1990, 29, 4335–4340 CrossRef CAS.
- M. Bartholoma, J. Valliant, K. P. Maresca, J. Babich and J. Zubieta, Chem. Commun., 2009, 493–512 RSC.
- F. L. Thorp-Greenwood, R. G. Balasingham and M. P. Coogan, J. Organomet. Chem., 2012, 714, 12–21 CrossRef CAS.
- R. G. Balasingham, M. P. Coogan and F. L. Thorp-Greenwood, Dalton Trans., 2011, 40, 11663–11674 RSC.
- V. Fernandez-Moreira, F. L. Thorp-Greenwood and M. P. Coogan, Chem. Commun., 2010, 46, 186–202 RSC.
- K. K.-W. Lo, A. W.-T. Choi and W. H.-T. Law, Dalton Trans., 2012, 41, 6021–6047 RSC.
- K. K.-W. Lo, K. Y. Zhang and S. P.-Y. Li, Eur. J. Inorg. Chem., 2011, 2011, 3551–3568 CrossRef CAS.
- K. K.-W. Lo, M.-W. Louie and K. Y. Zhang, Coord. Chem. Rev., 2010, 254, 2603–2622 CrossRef CAS.
- E. Baggaley, J. A. Weinstein and J. A. G. Williams, Coord. Chem. Rev., 2012, 256, 1762–1785 CrossRef CAS.
- E. V. Jensen and E. R. DeSombre, Science, 1973, 182, 126–134 CAS.
- G. G. J. M. Kuiper, E. Enmark, M. Pelto-Huikko, S. Nilsson and J.-A. Gustafsson, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 5925–5930 CrossRef CAS.
- C. Carmeci, D. A. Thompson, H. Z. Ring, U. Francke and R. J. Weigel, Genomics, 1997, 45, 607–617 CrossRef CAS.
- G. Notas, M. Kampa, V. Pelekanou and E. Castanas, Steroids, 2012, 77, 943–950 CrossRef CAS.
- S. Niro, E. Pereira, M.-A. Pelissier, R. Morfin and O. Hennebert, Steroids, 2012, 77, 542–551 CrossRef.
- E. J. Filardo, J. A. Quinn, A. R. Frackelton, Jr and K. I. Bland, Mol. Endocrinol., 2002, 16, 70–84 CrossRef CAS.
- S. Dong, S. Terasaka and R. Kiyama, Environ. Pollut., 2011, 159, 212–218 CrossRef CAS.
- S. P. Fraser, I. Ozerlat-Gunduz, R. Onkal, J. K. J. Diss, D. S. Latchman and M. B. A. Djamgoz, J. Cell. Physiol., 2010, 224, 527–539 CrossRef CAS.
- C. M. Revankar, D. F. Cimino, L. A. Sklar, J. B. Arterburn and E. R. Prossnitz, Science, 2005, 307, 1625–1630 CrossRef CAS.
- M. Maggiolini and D. Picard, J. Endocrinol., 2010, 204, 105–114 CrossRef CAS.
- E. R. Prossnitz, J. B. Arterburn and L. A. Sklar, Mol. Cell. Endocrinol., 2007, 265–266, 138–142 CrossRef CAS.
- K. Matsuda, H. Sakamoto, H. Mori, K. Hosokawa, A. Kawamura, M. Itose, M. Nishi, E. R. Prossnitz and M. Kawata, Neurosci. Lett., 2008, 441, 94–99 CrossRef CAS.
- H. Sakamoto, K.-i. Matsuda, K. Hosokawa, M. Nishi, J. F. Morris, E. R. Prossnitz and M. Kawata, Endocrinology, 2007, 148, 5842–5850 CrossRef CAS.
- M. Reisgys, F. Wust, R. Alberto, R. Schibli, P. A. Schubiger, H.-J. Pietzsch, H. Spies and B. Johannsen, Bioorg. Med. Chem. Lett., 1997, 7, 2243–2246 CrossRef CAS.
- R. J. Miksicek, K. E. Carlson, K.-J. Hwang and J. A. Katzenellenbogen, Mol. Endocrinol., 1995, 9, 592–604 CrossRef CAS.
- H. F. VanBrocklin, K. E. Carlson, J. A. Katzenellenbogen and M. J. Welch, J. Med. Chem., 1993, 36, 1619–1629 CrossRef CAS.
- J. Shani, A. Gazit, T. Livshitz and S. Biran, J. Med. Chem., 1985, 28, 1504–1511 CrossRef CAS.
- F. Benard, N. Ahmed, J.-M. Beauregard, J. Rousseau, A. Aliaga, C. Dubuc, E. Croteau and J. E. Lier, Eur. J. Nucl. Med. Mol. Imaging, 2008, 35, 1473–1479 CrossRef CAS.
- L. G. Luyt, H. M. Bigott, M. J. Welch and J. A. Katzenellenbogen, Bioorg. Med. Chem., 2003, 11, 4977–4989 CrossRef CAS.
- H. M. Bigott, E. Parent, L. G. Luyt, J. A. Katzenellenbogen and M. J. Welch, Bioconjugate Chem., 2005, 16, 255–264 CrossRef CAS.
- H. Enginar, P. Unak, F. Y. Lambrecht, F. Z. Biber Muftuler and E. I. Medine, In Vivo, 2008, 22, 93–100 CAS.
- P. M. Martin, H. P. Magdelenat, B. Benyahia, O. Rigaud and J. A. Katzenellenbogen, Cancer Res., 1983, 43, 4956–4965 CAS.
- M. Okamoto, S. Kobayashi, H. Ikeuchi, S. Yamada, K. Yamanouchi, K. Nagasawa, S. Maekawa, T. Kato and I. Shimizu, Steroids, 2012, 77, 845–849 CrossRef CAS.
- I. Jose, K. D. Deodhar, U. B. Desai and S. Bhattacharjee, J. Fluoresc., 2011, 21, 1171–1177 CrossRef CAS.
- B. Fisher, N. Gunduz, S. Zheng and E. A. Saffer, Cancer Res., 1982, 42, 540–549 CAS.
- E. L. Rickert, S. Oriana, C. Hartman-Frey, X. Long, T. T. Webb, K. P. Nephew and R. V. Weatherman, Bioconjugate Chem., 2010, 21, 903–910 CrossRef CAS.
- K. K.-W. Lo, K. H.-K. Tsang, K.-S. Sze, C.-K. Chung, T. K.-M. Lee, K. Y. Zhang, W.-K. Hui, C.-K. Li, J. S.-Y. Lau, D. C.-M. Ng and N. Zhu, Coord. Chem. Rev., 2007, 251, 2292–2310 CrossRef CAS.
- K. K.-W. Lo, T. K.-M. Lee, J. S.-Y. Lau, W.-L. Poon and S.-H. Cheng, Inorg. Chem., 2008, 47, 200–208 CrossRef CAS.
- K. K.-W. Lo, K. H.-K. Tsang and N. Zhu, Organometallics, 2006, 25, 3220–3227 CrossRef CAS.
- R. Masyuko, E. J. Lanni, J. V. Sweedler and P. W. Bohn, Analyst, 2013, 138, 1924–1939 RSC.
- T. A. Ternes, H. Andersen, D. Gilberg and M. Bonerz, Anal. Chem., 2002, 74, 3498–3504 CrossRef CAS.
- A. Nelson, Int. J. Women's Health, 2010, 2, 99–106 CAS.
- A. Vessieres, S. Top, C. Vaillant, D. Osella, J. Mornon and G. Jaouen, Angew. Chem., Int. Ed. Engl., 1992, 104, 790–792 CAS.
- J. B. Arterburn, C. Corona, K. V. Rao, K. E. Carlson and J. A. Katzenellenbogen, J. Org. Chem., 2003, 68, 7063–7070 CrossRef CAS.
- V. Novakova, P. Zimcik, M. Miletin, K. Kopecky and J. Ivincova, Tetrahedron Lett., 2010, 51, 1016–1018 CrossRef CAS.
- N. V. Sokolova and V. G. Nenajdenko, Chem. Heterocycl. Compd., 2012, 48, 903–906 CrossRef CAS.
- C. B. Anderson, A. B. S. Elliott, C. J. McAdam, K. C. Gordon and J. D. Crowley, Organometallics, 2013, 32, 788–797 CrossRef CAS.
- F. Wust, M. B. Skaddan, P. Leibnitz, H. Spies, J. A. Katzenellenbogen and B. Johannsen, Bioorg. Med. Chem., 1999, 7, 1827–1835 CrossRef CAS.
- C. Ramesh, B. J. Bryant, T. Nayak, C. M. Revankar, T. Anderson, K. E. Carlson, J. A. Katzenellenbogen, L. A. Sklar, J. P. Norenberg, E. R. Prossnitz and J. B. Arterburn, J. Am. Chem. Soc., 2006, 128, 14476–14477 CrossRef CAS.
- T. W. Spradau and J. A. Katzenellenbogen, Bioorg. Med. Chem. Lett., 1998, 8, 3235–3240 CrossRef CAS.
- K.-L. Dao, R. R. Sawant, J. A. Hendricks, V. Ronga, V. P. Torchilin and R. N. Hanson, Bioconjugate Chem., 2012, 23, 785–795 CrossRef CAS.
- M. Salmain, A. Vessières, G. Jaouen and I. S. Butler, Anal. Chem., 1991, 63, 2323–2329 CrossRef CAS.
- T. Y. Kim, A. B. S. Elliott, K. J. Shaffer, C. J. McAdam, K. C. Gordon and J. D. Crowley, Polyhedron, 2013, 52, 1391–1398 CrossRef CAS.
- A. Boulay, A. Seridi, C. Zedde, S. Ladeira, C. Picard, L. Maron and E. Benoist, Eur. J. Inorg. Chem., 2010, 5058–5062 CAS.
- M. Obata, A. Kitamura, A. Mori, C. Kameyama, J. A. Czaplewska, R. Tanaka, I. Kinoshita, T. Kusumoto, H. Hashimoto, M. Harada, Y. Mikata, T. Funabiki and S. Yano, Dalton Trans., 2008, 3292–3300 RSC.
- R. Burai, C. Ramesh, T. K. Nayak, M. K. Dennis, B. K. Bryant, E. R. Prossnitz and J. B. Arterburn, PLoS One, 2012, 7, e46861 CAS.
- E. A. Hillard, A. Vessieres, S. Top, P. Pigeon, K. Kowalski, M. Huche and G. Jaouen, J. Organomet. Chem., 2007, 692, 1315–1326 CrossRef CAS.
- D.-H. Han, H. Tachibana and K. Yamada, In Vitro Cell. Dev. Biol.: Anim., 2001, 37, 275–282 CAS.
- A. Gupta, D. Mandal, Y. Ahmadibeni, K. Parang and G. Bothun, Eur. Biophys. J., 2011, 40, 727–736 CrossRef CAS.
- D. Colin, A. Lancon, D. Delmas, G. Lizard, J. Abrossinow, E. Kahn, B. Jannin and N. Latruffe, Biochimie, 2008, 90, 1674–1684 CrossRef CAS.
- M. D. Bartholoma, A. R. Vortherms, S. Hillier, J. Joyal, J. Babich, R. P. Doyle and J. Zubieta, Dalton Trans., 2011, 40, 6216–6225 RSC.
- C. Sandt, J. Frederick and P. Dumas, J. Biophotonics, 2013, 6, 60–72 CrossRef CAS.
- T. Dieing, O. Hollricher and J. Toporski, Confocal Raman Spectroscopy, Springer, 2010 Search PubMed.
- E. Gazi, J. Dwyer, N. P. Lockyer, J. Miyan, P. Gardner, C. Hart, M. Brown and N. W. Clarke, Biopolymers, 2005, 77, 18–30 CrossRef CAS.
- V. Zinchuk and O. Grossenbacher-Zinchuk, Curr. Protoc. Cell Biol., 2011, ch. 4, Unit4 19 Search PubMed.
- Q. Li, A. Lau, T. J. Morris, L. Guo, C. B. Fordyce and E. F. Stanley, J. Neurosci., 2004, 24, 4070–4081 CrossRef CAS.
- J. M. Hall, J. F. Couse and K. S. Korach, J. Biol. Chem., 2001, 276, 36869–36872 CrossRef CAS.
- K. P. Yang and N. A. Samaan, Cancer Res., 1983, 43, 3534–3538 CAS.
- C. M. Revankar, H. D. Mitchell, A. S. Field, R. Burai, C. Corona, C. Ramesh, L. A. Sklar, J. B. Arterburn and E. R. Prossnitz, ACS Chem. Biol., 2007, 2, 536–544 CrossRef CAS.
- C. Otto, B. Rohde-Schulz, G. Schwarz, I. Fuchs, M. Klewer, D. Brittain, G. Langer, B. Bader, K. Prelle, R. Nubbemeyer and K.-H. Fritzemeier, Endocrinology, 2008, 149, 4846–4856 CrossRef CAS.
- D. J. Minick, J. H. Frenz, M. A. Patrick and D. A. Brent, J. Med. Chem., 1988, 31, 1923–1933 CrossRef CAS.
- M. G. Pomper, H. VanBrocklin, A. M. Thieme, R. D. Thomas, D. O. Kiesewetter, K. E. Carlson, C. J. Mathias, M. J. Welch and J. A. Katzenellenbogen, J. Med. Chem., 1990, 33, 3143–3155 CrossRef CAS.
- P. Dumas, F. Polack, B. Lagarde, O. Chubar, J. L. Giorgetta and S. Lefrancois, Infrared Phys. Technol., 2006, 49, 152–160 CAS.
- A. Giuliani, F. Jamme, V. Rouam, F. Wien, J.-L. Giorgetta, B. Lagarde, O. Chubar, S. Bac, I. Yao, S. Rey, C. Herbeaux, J.-L. Marlats, D. Zerbib, F. Polack and M. Refregiers, J. Synchrotron Radiat., 2009, 16, 835–841 CrossRef.
- F. Jamme, S. Villette, A. Giuliani, V. Rouam, F. Wien, B. Lagarde and M. Refregiers, Microsc. Microanal., 2010, 16, 507–514 CrossRef CAS.
- A. Dazzi, M. Reading, P. Rui and K. Kjoller, Patent WO/2008/143817, 2008.
- A. Dazzi, R. Prazeres, F. Glotin and J.-M. Ortega, Infrared Phys. Technol., 2006, 49, 113–121 CAS.
- A. Dazzi, R. Prazeres, F. Glotin and J.-M. Ortega, Ultramicroscopy, 2007, 107, 1194–1200 CrossRef CAS.
- A. Dazzi, R. Prazeres, F. Glotin, J.-M. Ortega, M. Al-Sawaftah and M. de Frutos, Ultramicroscopy, 2008, 108, 635–641 CrossRef CAS.
- A. Dazzi, F. Glotin and R. Carminati, J. Appl. Phys., 2010, 107, 124519 CrossRef.
- C. Mayet, A. Deniset, R. Prazeres, J.-M. Ortega and A. Dazzi, Biotechnol. Adv., 2013, 31, 369–374 CrossRef CAS.
- A. Dazzi and C. Policar, in Biointerface Characterization by Advanced IR Spectroscopy, ed. C.-M. Pradier and Y. Chabal, Elsevier, 2011, pp. 245–278 Search PubMed.
- C. J. Hirschmugl and K. M. Gough, Appl. Spectrosc., 2012, 66, 475–491 CrossRef CAS.
- M. J. Nasse, M. J. Walsh, E. C. Mattson, R. Reininger, A. Kajdacsy-Balla, V. Macias, R. Bhargava and C. J. Hirschmugl, Nat. Methods, 2011, 8, 413–416 CrossRef CAS.
- E. C. Mattson, M. Unger, B. Manandhar, Z. Alavi and C. J. Hirschmugl, J. Phys.: Conf. Ser., 2013, 425, 142001 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3an00807j |
| This journal is © The Royal Society of Chemistry 2013 |