 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High-throughput analysis of drugs in biological fluids by desorption electrospray ionizationmass spectrometry coupled with thin liquid membrane extraction
Cecilie
Rosting
a,
Stig
Pedersen-Bjergaard
ab,
Steen Honoré
Hansen
b and
Christian
Janfelt
*b
aSchool of Pharmacy, University of Oslo, Norway
bDepartment of Pharmacy, University of Copenhagen, Denmark. E-mail: christian.janfelt@sund.ku.dk; Fax: +45 35 33 60 30; Tel: +45 35 33 65 57
First published on 24th July 2013
Abstract
Biological fluids such as urine, saliva and whole blood were analyzed for contents of drugs by a new combination of desorption electrospray ionizationmass spectrometry (DESI-MS) and thin liquid membrane extraction (TLME). Analytes from the sample were extracted into a thin liquid membrane of hexadecane deposited on a porous Teflon membrane, from which they were subsequently analyzed directly by DESI. The total analysis time was 15 minutes for analysis of several samples with a potential analysis time of less than a minute per sample. Thanks to the pre-concentration and sample clean-up built into the method, methadone was detected in urine in full-scan mode with an LOD of 4 ng mL−1, while amitriptyline, nortriptyline and pethidine showed LODs of 17 ng mL−1. Quantification was possible for several basic drugs using one common internal standard, providing relative accuracies in the range of 10–30%. A reliability test was performed on 20 samples with methadone, amitriptyline, nortriptyline and pethidine in urine, showing that none of the samples having concentrations above the LOD were missed and no false positives were found. Diphenhydramine and one of its metabolites were detected in authentic samples of urine and saliva, and methadone was detected from a whole-blood sample spiked to a concentration of 100 ng mL−1. The method has several advantages, such as extremely low price in consumables, the possibility of fast analysis of very crude biofluids such as whole blood and the potential for a very high sample throughput.
Introduction
Chemical analysis of drug substances in biological fluids is performed in several highly important disciplines like therapeutic drug monitoring,1 clinical research,2 forensic science,3 and doping in sports.4 Blood, urine, and saliva are the most important types of samples collected for this purpose.5 Prior to chemical analysis, some type of sample preparation is performed, typically either by solid-phase extraction (SPE), protein precipitation (PP), or liquid–liquid extraction (LLE).6 The chemical analysis is in most cases performed by liquid chromatography (LC) coupled with mass spectrometry (MS) or with tandem mass spectrometry (MS/MS).7LC-MS and LC-MS/MS provide high specificity, accuracy, and precision, but the methods are often relatively time-consuming. This may limit the sample through-put to some degree, and alternative strategies are generally of high interest.One strategy to increase the sample through-put is using desorption electrospray ionizationmass spectrometry (DESI-MS).8DESI-MS has previously been demonstrated for direct analysis of dried spots of dilute raw urine,9 but in most other cases where DESI was used for urine analysis, the approach has been to couple some extraction technique with DESI, in recognition of the considerable matrix effects associated with urine analysis, even when DESI is applied as an analysis technique. DESI has thus been applied for analysis of various biological fluids in conjunction with solid phase extraction (SPE),10,11solid phase microextraction (SPME),12 liquid–liquid extraction (LLE),13 thin film microextraction (TFME)14 and liquid phase microextraction (LPME).15
SPE DESI can be performed by making an ordinary solid phase extraction, depositing the eluate on a surface for evaporation and subsequently analyzing the sample spot by DESI, as done by Lin et al. who developed a method for screening of clenbuterol in urine by SPE-DESI.11 Alternatively, it can be done by performing the DESI analysis directly on the SPE material, as accomplished in an automated fashion by Takáts and co-workers.10 More recently, Takáts and co-workers demonstrated a similar approach using thin-film microextraction (TFME) blades, benefiting from their low film thickness and flat geometry which make them suitable for coupling to DESI in e.g. analysis of wastewater.14
Another way to couple extraction techniques with mass spectrometry without use of chromatography is the coupling of microextraction by packed sorbent (MEPS) and electrospray ionization from a microchip as presented by Ketola and co-workers in 2012 for analysis of drugs in urine samples.16 A quite similar approach was used in our introduction of the capillary spray cell which is a simple electrospray device well-suited for analysis of crude plant extracts.17
Cooks and co-workers combined the extraction and spray device into one unit in the introduction of PaperSpray.18 With PaperSpray the sample is applied to a piece of paper, e.g. by wiping or deposition, the paper is cut in a triangular shape, and an electrospray can be formed from the apex of the paper by application of a high voltage and a drop of methanol applied to the paper. The extraction of the sample and the ionization thus take place virtually simultaneously.
We have previously coupled liquid phase microextraction (LPME) with DESI in a 3-phase setup where an aqueous sample was extracted over a hydrophobic liquid membrane into an aqueous acceptor phase, providing extremely clean and highly pre-concentrated extracts.15 The amount of extract, a few microliters, was deposited on a surface and analyzed by DESI. The method provided sensitive detection of drugs in urine, but suffered from relatively long drying times after deposition of the aqueous extracts on the surface. With the present DESI approach measurements were performed directly from a hydrophobic liquid membrane, and this approach was termed thin liquid membrane extraction (TLME). The analytes were thus analyzed directly from a liquid membrane sustained in the pores of a porous Teflon membrane. The concept was somewhat similar to the work by Chen and Miao on direct DESI analysis of liquid samples,19 but in the present work the analytes were analyzed from an organic liquid rather than an aqueous liquid. Besides the reduced total analysis time, the detection of the drugs directly from the thin liquid membrane has the advantage that the method does not suffer from different drying patterns of different compounds, thus making the choice of an internal standard less critical.
Experimental
Chemicals
LC-MS grade methanol was purchased from VWR International (Herlev, Denmark), and water was prepared with a Millipore Direct-Q3 UV system (Billerica, MA, USA). Isotopically labeled amitriptyline-d6 and diphenhydramine-d5 were purchased from Qmx Laboratories (Essex, UK). All other chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA).Thin liquid membrane extraction (TLME)
The principle of the thin liquid membrane extraction setup which was used is illustrated in Fig. 1. An organic solvent immiscible with water was deposited on a porous Teflon membrane, forming a thin liquid membrane. The thin liquid membrane served as the bottom of a well to which the sample was added for extraction; a lid was added in order to allow for agitation during extraction. During the extraction the relatively non-polar analytes were dissolved in the thin liquid membrane, from which they were later desorbed and analyzed by DESI. The DESI analysis was performed either on the front side of the thin liquid membrane, which has been in direct contact with the sample, or on the backside of the thin liquid membrane. The latter was feasible because the analytes diffused across the thin liquid membrane and were equally distributed over the cross-section.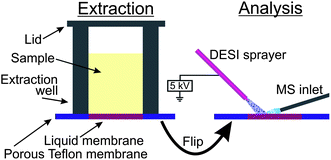 | ||
| Fig. 1 The principle of TLME-DESI. | ||
Our implementation of the method is shown in Fig. 2. We constructed a three layer sandwich in aluminium, comprising a frame to fix the supporting Teflon membrane, a plastic insert to add the sample cells (4 × 2 wells, 6 mm diameter, 10 mm height, total volume 280 μL), and a lid to seal the sample cells during agitation. As a supporting membrane we used a porous Teflon membrane (200 μm thick, medium pore size of 7 μm, pore volume of 36%, purchased from Berghof (Eningen, Germany)). 1.5 μL of hexadecane was added by a pipette, forming the thin liquid membrane for each sample cell. 150 μL of the sample (aqueous, urine, whole blood or saliva) was pipetted into the sample cells, 40 μL of 500 ng mL−1 amitriptyline-d6 was added as an internal standard, and 20 μL of 1 M NaOH was added in order to obtain a pH > 7 and force the basic drugs from the hydrophilic sample into the lipophilic thin liquid membrane. The extraction unit was sealed with a lid and placed on a vortex mixer (VWR VV3, VWR, Herlev, Denmark) for 10 minutes. Subsequently, the samples were discarded, and the Teflon membrane was mounted with double-sided tape on a microscope glass slide, with the backside of the thin liquid membrane facing up. In this way, DESI-MS was performed from a part of the thin liquid membrane which has not been in contact with the sample. This was advantageous in urine analysis and of crucial importance in whole blood analysis. The Teflon membrane was ready for DESI-MS analysis immediately after extraction. The analytes were thus desorbed and ionized directly from the thin (organic) liquid membrane without any need for drying.
 | ||
| Fig. 2 Implementation of TLME-DESI. (a) 1.5 μL hexadecane deposited on a porous Teflon membrane. (b) Sample cells are fastened on top of the liquid membranes, and the sample, NaOH and internal standard are pipetted into the sample cells. (c) The lid is fastened, sealing the sample cells, and the assembly is agitated for 10 minutes on a vortex mixer. (d) DESI-MS analysis of the Teflon membrane. | ||
DESI-MS analysis
DESI-MS analysis was performed on an LTQ XL Linear Ion Trap Mass Spectrometer (Thermo Fisher Scientific, San Jose, CA, USA) equipped with a custom-built DESI imaging ion source, based on a motorized microscope stage from Märzhäuser Wetzlar (Wetzlar, Germany) and controlled with an in-house written software program, as described in detail elsewhere.20 The spray solvent was methanol with 1% formic acid, and the flow rate was 3 μL min−1. The nebulizer gas pressure was 7 bar, and 5 kV potential was applied to the sprayer. Thanks to the good properties of porous Teflon in DESI analysis,21 the geometric parameters were not very crucial; the spray angle was 55°, and the spray–inlet distance and spray–surface distance were around 3 mm and 1.5 mm, respectively. Analysis was made in full-scan and positive ion mode with a scan range of m/z 150–600. The injection time was 100 ms, and 2 micro-scans were used. The surface was scanned at a speed of 1 mm s−1, resulting in a total analysis time of 45 seconds for four samples, i.e. about 11 seconds per sample. It was possible to scan the four hexadecane spots several times, e.g. in rows separated by 0.5 mm space, in order to obtain more measurements and improve the statistics of the DESI experiment, but in practice one scan per sample was typically enough to provide results of adequate quality.The results were analyzed in Xcalibur 2.0 (Thermo Fisher Scientific). Extracted ion chronograms were generated for the relevant ions, and areas were integrated for each sample spot. Limits of detection (LOD) and quantification were based on the ratios between the analyte and the internal standard, using the ratio of the MS intensities in LOD experiments (as described in further detail in the following section), and for quantitation the ratios of the areas were used.
LC-MS analysis
LC-MS was applied on matrix-free aqueous samples in order to characterize the thin liquid membrane extraction separately from the DESI analysis, e.g. in optimization of the extraction time, by measuring the depletion of analyte in the donor phase. 80 μL of the sample was transferred to a vial after TLME and 5 μL of 5% formic acid was added in order to acidify the sample. The sample was analyzed on an LC-MS system (Agilent 1100 LC, and Thermo LCQ Deca XP Plus ion trap MS) equipped with a Phenomenex Synergi 4u Fusion-RP 80 column (100 × 2 mm, 4 μm). The injection volume was 5 μL, and the column temperature was 30 °C. Mobile phase A was Milli-Q water with 0.1% formic acid, and mobile phase B was methanol with 0.1% formic acid. The flow rate was 250 μL min−1, and gradient elution was used as follows: 0–4 minutes: rising from 5% B to 90% B; 4–5 minutes: 90% B isocratic; 5.0–5.1 minutes: ramping down to 5% B; 5.1–9 minutes: 5% B isocratic. The mass spectrometer was operated in positive ion and full-scan mode with a scan range of m/z 50–600. A three point calibration curve was made with concentrations in the range of 100–1000 ng mL−1, and the concentrations of the samples were calculated on the basis of integrated areas in the extracted ion chromatograms of the different compounds. 8 parallel extractions were made for each time point, and each extraction was analyzed separately by LC-MS.Spiked urine samples
Morning urine was collected from three healthy volunteers, mixed and used to prepare standard urine solution for calibration curves. For the reliability test, morning urine from a fourth healthy person was spiked with various amounts of the four drugsmethadone, amitriptyline, nortriptyline and pethidine at concentrations of 100–1000 ng mL−1.Spiked blood samples
A total of 20 mL of whole blood was collected in heparin tubes at the Blood Bank of Rigshospitalet (Copenhagen, Denmark). The blood was stored at 5 °C and spiked with drugs at various concentrations and analyzed on the following day.Diphenhydramine in authentic biological fluids
The prescribed dose of 2 tablets of Tylenol PM (McNeil Healthcare, PA, USA) containing a total of 50 mg of diphenhydramine and 1000 mg of paracetamol was taken by a 76 kg male volunteer. After two hours, samples were taken of blood, saliva and urine. A total of 500 μL of blood was collected in small heparin tubes (Microvette 100 LH, Sarstedt, Nümbrecht, Germany). The samples were analyzed within the first hour after they were taken. Isotope labeled diphenhydramine-d5 was added as an internal standard in all analyses to a concentration of 100 ng mL−1.Results and discussion
Optimizing the experimental parameters
A number of different hydrophobic compounds were tested as the thin liquid membrane, including 1-octanol, toluene, 2-nitrophenyloctylether, ethyl dodecanoate, dodecane, 1-bromododecane, 2-undecanone, pentylbenzene, tetrachlorophenylpolydimethylsiloxane, polyphenylmethylsiloxane, poly-3,3,3-trifluoropropylmethylsiloxane, and hexadecane. In general, pentylbenzene, dodecane, 1-bromododecane, 2-undecanone and in particular hexadecane were well-suited as liquid membranes. Several factors are crucial in the selection of the liquid membrane. It turns out that compounds such as 1-octanol and 2-octanone are not soaked into the supporting porous Teflon membrane, most likely due to the very hydrophobic nature of Teflon, and form a bubble on the surface instead. Hexadecane turned out to be ideal as a liquid membrane, as it distributes nicely into the pores of the Teflon membrane and does not readily evaporate. It thus appears that the polarity of the liquid membrane is of greater importance than surface tension for distribution in the pores, since e.g.1-octanol and hexadecane show different behaviors despite very similar values of surface tension (27.10 mN m−1 and 27.05 mN m−1, respectively). Furthermore, as hexadecane is highly non-polar, it does not interfere with the DESI experiment, even when we were spraying directly on the liquid membrane, since non-polar solvents are not ionized by the electrospray. In our choice of the liquid membrane we have not had a case where one of the tested membranes were too viscous; we do however expect that very viscous fluids will be less suited as membranes, since high viscosity will limit the diffusion through the liquid membrane which is of great importance during the extraction.Likewise, a number of different spray solvents were tested, involving methanol or acetonitrile with water in various mixing ratios and with and without addition of formic acid. The optimal spray solvent was found to be methanol with 1% formic acid. It is most likely of importance that methanol – although not entirely miscible with hexadecane – mixes better with the hydrophobic membrane than does e.g. a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 methanol–water mixture, and thus enables better access to the extracted analytes for desorption and ionization by DESI.
:1 methanol–water mixture, and thus enables better access to the extracted analytes for desorption and ionization by DESI.
As seen, the choices of the supporting material, liquid membrane and DESI spray solvent must be made together, since the change of the items will influence what is optimal for the others. One way, for example, to address the problem that 1-octanol does not distribute into the pores of a Teflon surface is to use porous polypropylene instead, which thus enables the use of more polar liquid membranes.
In an initial characterization of the thin liquid membrane extraction concept, 1 μg mL−1 matrix-free aqueous solutions of methadone, amitriptyline, nortriptyline and pethidine were extracted into the liquid membrane, using different extraction times, and the depletion of the samples was analyzed by LC-MS. Fig. 3 shows the depletion of the four drug substances. As seen, already after 10 minutes there was less than 20% of the initial amount of methadone, amitriptyline and nortriptyline left in the sample, while pethidine rapidly stabilized at 75%. In other words, the extraction recovery was above 80% for methadone, amitriptyline, and nortriptyline (10 minutes extraction), while for pethidine the recovery was 25%. This was in good agreement with logP values of the first three compounds of 3.9, 4.9 and 4.5, respectively, while pethidine is more polar with a logP value of 2.7 and thus less likely to be extracted into the highly hydrophobic liquid membrane of hexadecane. Similar experiments were conducted with morphine which due to its very hydrophilic nature (logP = 0.89) was not extracted into the liquid membrane.
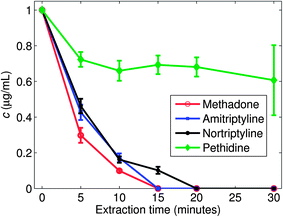 | ||
| Fig. 3 Depletion of different drugs at different times in the sample phase of the TLME-DESI setup, as investigated by LC-MS. The initial concentration was 1000 ng mL−1. | ||
Examples of the data generated with a TLME-DESI experiment are shown in Fig. 4 and 5. Fig. 4 shows the DESI mass spectra recorded from the liquid membrane after extraction of a urine sample spiked to a concentration of 25 ng mL−1 of the four drug substances and 100 ng mL−1 of the internal standard. Due to the high boiling point of hexadecane (281 °C) the liquid membrane was intact during the entire DESI experiment, so the analytes were desorbed and ionized directly from the hexadecane membrane and not from a dry Teflon membrane. A significant improvement was obtained by analyzing the liquid membrane from the backside (Fig. 4a) compared to the front side (Fig. 4b), as seen by higher intensities and fewer peaks in the background. The front side was in contact with the biological sample during extraction, and some fouling of the membrane surface by the sample matrix potentially occurred. However, due to the thin nature of the liquid membrane, the drug substances rapidly diffused and distributed equally over the cross-section of the membrane. Thus, the analytes were detected efficiently on the backside of the membrane, without potential interferences due to membrane fouling or other sample matrix related problems.
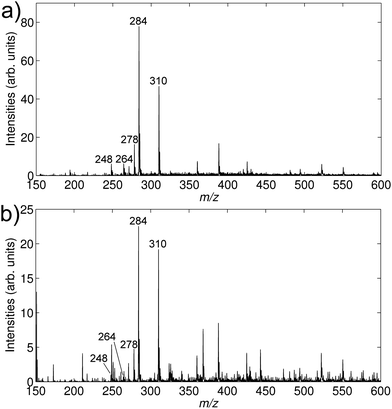 | ||
| Fig. 4 DESI mass spectra from an extracted urine sample spiked to concentrations of 25 ng mL−1 of amitriptyline (m/z 278), nortriptyline (m/z 264), methadone (m/z 310) and pethidine (m/z 248). Isotope labeled amitriptyline-d6 (m/z 284) was added as an internal standard to a concentration of 100 ng mL−1. (a) DESI-MSspectrum of the backside of the Teflon membrane. (b) DESI-MSspectrum of the front side of the Teflon membrane. | ||
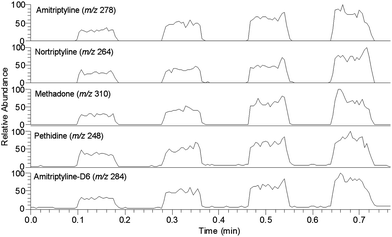 | ||
| Fig. 5 Extracted ion chronograms of amitriptyline (m/z 278), nortriptyline (m/z 264), methadone (m/z 310) and pethidine (m/z 248), all present at 300 ng mL−1 in a spiked urine sample, and amitriptyline-d6 (m/z 284) at 100 ng mL−1. The x-axis represents the scan time, as the Teflon membrane is scanned below the DESI sprayer, spot by spot. | ||
Fig. 5 shows an example of an extracted ion chronogram (EIC) from an extraction experiment on a urine sample spiked with 300 ng mL−1 of the four drugs and 100 ng mL−1 of the internal standard. The experiment was made in four neighboring sample cells. As observed, the signal differs significantly between the four sample cells, owing to differences in the extraction or detection. The method was therefore highly dependent on the presence of an internal standard. Fortunately, it turned out that the mutual differences between the four sample cells were well reproduced for all five extracted compounds, and the method was therefore reliable when used with an internal standard (discussed below).
Establishment of limits of detection (LOD)
The LOD was established in spiked urine samples. The definition was based on the ratio of the average MS signals over a sample spot (as read in the EIC) between the analyte and the internal standard. This ratio was measured on a blank urine sample, and we defined the LOD as the concentration which would result in a ratio five times higher than that of the blank sample. In this way, we obtained limits of detection in urine of 4 ng mL−1 for methadone and 17 ng mL−1 for amitriptyline, nortriptyline and pethidine. We performed a reliability test of 20 samples with the four compounds together spiked at concentrations ranging from 0–500 ng mL−1, for each sample four extractions were performed simultaneously. The compounds were detected in all samples where they were present at concentrations above the LOD, and in a few samples where they were present at concentrations below the LOD (e.g. 10 ng mL−1 of amitriptyline). More importantly, the reliability test showed no false positives, i.e. in no case was a signal detected from a compound in a sample which did not contain the given compound. The LODs reported in this work were as expected somewhat higher than those reported with chromatographic techniques coupled with mass spectrometry.22,23 However, the current LODs with TLME-DESI were fully acceptable for work with real samples.24 In addition, we expect to improve our LODs in the near future by implementing a more sensitive mass spectrometer.The quantitative performance of TLME-DESI
Calibration curves were made for the four compounds, using amitriptyline-d6 as an internal standard, by analyzing urine samples spiked to concentrations of 50, 100, 200 and 300 ng mL−1, and plotting the ratio between the area of the signal from the individual compounds and the area of the internal standard. The calibration curves are shown in Fig. 6 with error bars indicating the standard deviations between the four extractions; the response from methadone, which also had the lowest LOD of the four compounds, has been divided by 10 in order to fit the signals in the same figure. As seen, good linearities were obtained for all four compounds (r2 ranged between 0.989 and 0.999), also those which are structurally different from the internal standard. The figure thus shows that an isotopically labeled internal standard was not strictly necessary for the method, although amitriptyline whose deuterated analogue was used as an internal standard showed slightly lower standard deviations (RSD < 5.0%) than the others (RSD 5.0–32.9%).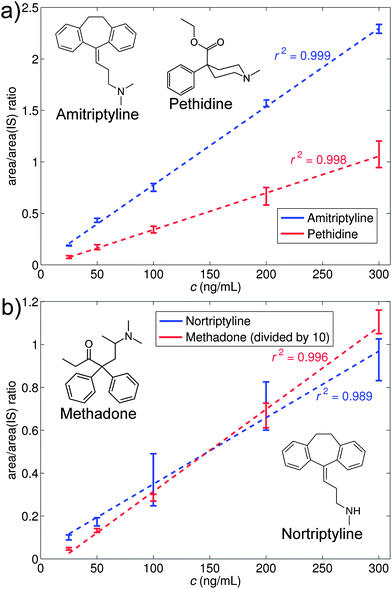 | ||
| Fig. 6 Calibration curves for urine samples spiked with (a) amitriptyline and pethidine, and (b) nortriptyline and methadone. Isotope labeled amitriptyline-d6 was used as an internal standard at a concentration of 100 ng mL−1. | ||
The calibration curves were used to quantify eight urine samples spiked with the four compounds at various concentrations. If the samples showed contents of any of the analytes (i.e. S/N ratio > 5, relative to a blank sample), the amount was quantified by means of the calibration curve. It should be pointed out that different urines were used for the calibration curve and the spiked samples in order to show a more realistic use of the method. The result is shown in Table 1 which presents the spiked amounts together with the detected amounts. The compounds were in general detected with a deviation of 11% or less for amitriptyline and of 33% or less for the three other compounds. It appeared that the accuracy for amitriptyline was superior due to the use of deuterated amitriptyline as an internal standard. Yet, it is remarkable that we, given the variations within the individual extractions as seen in Fig. 5, can work quantitatively using internal standards which are neither isotope labeled nor structure analogues. This is in contrast to our previous LPME-DESI approach which was based on LPME extraction to an acceptor solution present in a hollow fibre.15 The LPME extract was deposited on a surface, evaporated and analyzed by DESI. This meant that the internal standard had to mimic the analyte behavior with respect to extraction into the acceptor phase, evaporation on the surface and desorption and ionization in the DESI experiment. In particular the evaporation step turned out to be critical as different compounds had different evaporation patterns, thus requiring quantitative analysis to be made with isotope labeled compounds or structure analogues as internal standards. In the current work, by analyzing the compounds while they were dissolved in the liquid membrane, we thus avoided the evaporation step which added extra uncertainty and complexity to the analysis.
| Test | Amitriptyline | Methadone | Nortriptyline | Pethidine | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| True | Det. | %dev. | True | Det. | %dev. | True | Det. | %dev. | True | Det. | %dev. | |
| 1 | 0 | 0 | 0 | 50 | 53 | 6 | 200 | 188 | −6 | 0 | 0 | 0 |
| 2 | 150 | 133 | −11 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 3 | 200 | 187 | −7 | 150 | 111 | −26 | 70 | 61 | −13 | 100 | 67 | −33 |
| 4 | 90 | 85 | −6 | 0 | 0 | 0 | 0 | 0 | 0 | 150 | 100 | −33 |
| 5 | 0 | 0 | 0 | 0 | 0 | 0 | 100 | 74 | −26 | 0 | 0 | 0 |
| 6 | 50 | 55 | 10 | 250 | 183 | −27 | 0 | 0 | 0 | 50 | 43 | −14 |
| 7 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 8 | 0 | 0 | 0 | 90 | 67 | −26 | 0 | 0 | 0 | 0 | 0 | 0 |
TLME-DESI on authentic samples of biofluids
In order to complement the results on the spiked urine samples, the method was also tested on authentic samples based on the intake of two tablets of the over-the-counter drugTylenol PM which contain 500 mg of paracetamol (acetaminophen) and 25 mg of diphenhydramine each. While paracetamol with its logP value of 0.46 was not extracted with the present setup, diphenhydramine with its logP value of 3.3 was well suited for analysis.
Samples were taken of urine and saliva two hours after oral administration of the drug and extracted and analyzed as described in the Experimental section, using deuterated diphenhydramine as an internal standard. The mass spectra of the urine and saliva samples are shown in Fig. 7a and b. Diphenhydramine was easily observed at m/z 256, and moreover a clear signal was seen at m/z 242 from the metabolite nordiphenhydramine. The relative abundance of nordiphenhydramine was highest in the urine sample as expected. The abundant signal at m/z 261 was from the deuterated internal standard. Examples of extracted ion chronograms of the diphenhydramine signal in the samples are shown in Fig. 7c and d, respectively. A calibration curve was generated in urine for diphenhydramine using the same procedure as above, and the concentration of diphenhydramine in the urine sample was found to be 126 ng mL−1.
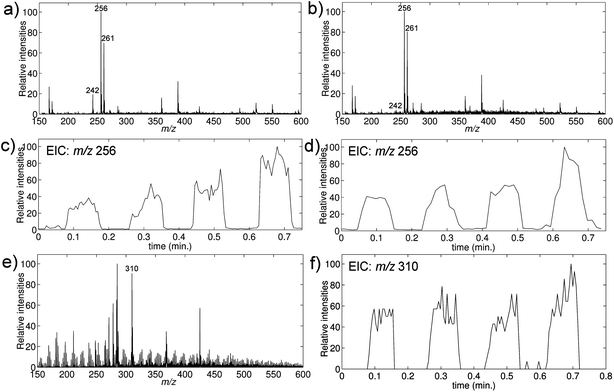 | ||
| Fig. 7 TLME-DESI analysis of biological samples. (a–d) Analyses of authentic samples of diphenhydramine in urine and saliva; the samples were obtained two hours after oral administration of 50 mg of diphenhydramine. (e and f) Analysis of a whole-blood sample spiked with methadone to a concentration of 100 ng mL−1. The mass spectra show the presence of diphenhydramine (m/z 256), the internal standard diphenhydramine-d5 (m/z 261), the metabolite nordiphenhydramine (m/z 342) and methadone (m/z 310). (a) DESImass spectrum of an extracted urine sample. (b) DESImass spectrum of an extracted saliva sample. (c) Extracted ion chronogram of diphenhydramine in urine. (d) Extracted ion chronogram of diphenhydramine in saliva. (e) DESImass spectrum of an extracted spiked whole-blood sample. (f) Extracted ion chronogram of methadone in spiked whole-blood. | ||
Furthermore, a blood sample from a 10 mL heparin tube received from the blood bank of Rigshospitalet was spiked to a concentration of 100 ng mL−1 of methadone, extracted and analyzed. The DESImass spectrum in Fig. 7e shows an intense signal at m/z 310 from methadone, and the EIC in Fig. 7f shows very certain detection of methadone in the extracted blood sample with methadone concentrations in the lower end of the therapeutic range (which is 100–500 ng mL−1 (ref. 24)).
Compared to solid phase extraction, the LPME-DESI method is extremely cheap in consumables, the only consumables being the porous Teflon membrane and hexadecane. With an average consumption of 1.3 cm2 of the Teflon membrane per sample with our present 8 sample cell setup, the Teflon membrane represents a cost of 0.021 EUR per sample while the price of hexadecane is negligible with a consumption of 1.5 μL per sample.
Conclusion
We have presented a new concept integrating thin liquid membrane extraction (TLME) and desorption electrospray ionizationmass spectrometry (DESI-MS), which allows analysis of samples of biological fluids in a very short time and at an extremely low cost. The extraction procedure provided significant sample clean-up as well as a pre-concentration of the analyte into a thin liquid membrane, and DESI-MS was accomplished directly from this liquid membrane. In our proof-of-principle implementation we extracted 150 μL of the sample into 1.5 μL of hexadecane as the thin liquid membrane, but the dimensions of the extraction cells, and thus the sample volume, can be adjusted to the relevant application. Likewise, with our setup we were analyzing 8 samples simultaneously, but the number could easily be much larger. We have demonstrated TLME-DESI-MS of several drug substances at clinically relevant concentrations, and with limits of detection down to the low ng mL−1 level after extraction for 10 minutes. We have applied an ion trap mass spectrometer in full scan mode, but it is likely that the quantitative performance of the method would benefit greatly from a triple quadrupole mass spectrometer running in MRM mode, as applied e.g. in paper spray experiments.25The detection time was approximately 11 seconds per sample, making an average total analysis time of less than a minute per sample (including sample handling, extraction and analysis) realistic for a larger set of samples. We have shown that TLME-DESI can be used for identification and quantification, and is compatible with a number of different biological fluids such as urine, saliva and whole-blood. With hexadecane as the liquid membrane, only non-polar drugs were extracted and detected, and consequently this membrane provided very efficient sample clean-up. Work is in progress to implement other types of liquid membranes for more polar substances, without sacrificing selectivity and sample clean-up.
Acknowledgements
Support from the Carlsberg Foundation and The Danish Council for Independent Research | Natural Sciences is gratefully acknowledged.References
- J. E. Adaway and B. G. Keevil, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2012, 883–884, 33–49 CrossRef CAS.
- M. Himmelsbach, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2012, 883–884, 3–17 CrossRef CAS.
- A. Polettini, Chromatographic Science Series, in Handbook of HPLC, 2011, vol. 101, pp. 661–682 Search PubMed.
- K. Deventer, K. Roels, F. T. Delbeke and P. Eenoo, Anal. Bioanal. Chem., 2011, 401, 421–432 CrossRef CAS.
- Y. Huang, R. Shi, W. Gee and R. Bonderud, Bioanalysis, 2012, 4, 1919–1931 CrossRef CAS.
- H. Kataoka, K. Saito and A. Yokoyama, in Handbook of Sample Preparation, ed. J. Pawliszyn and H. L. Lord, 2010, pp. 285–311 Search PubMed.
- W. D. van Dongen and W. M. A. Niessen, Bioanalysis, 2012, 4, 2391–2399 CrossRef CAS.
- Z. Takats, J. M. Wiseman, B. Gologan and R. G. Cooks, Science, 2004, 306, 471–473 CrossRef CAS.
- H. Chen, Z. Pan, N. Talaty, D. Raftery and R. G. Cooks, Rapid Commun. Mass Spectrom., 2006, 20, 1577–1584 CrossRef CAS.
- J. Denes, M. Katona, A. Hosszu, N. Czuczy and Z. Takats, Anal. Chem., 2009, 81, 1669–1675 CrossRef CAS.
- Z. Q. Lin, S. C. Zhang, M. X. Zhao, C. D. Yang, D. P. Chen and X. R. Zhang, Rapid Commun. Mass Spectrom., 2008, 22, 1882–1888 CrossRef CAS.
- J. H. Kennedy, C. Aurand, R. Shirey, B. C. Laughlin and J. M. Wiseman, Anal. Chem., 2010, 82, 7502–7508 CrossRef CAS.
- T. J. Kauppila, N. Talaty, T. Kuuranne, T. Kotiaho, R. Kostiainen and R. G. Cooks, Analyst, 2007, 132, 868–875 RSC.
- N. Strittmatter, R.-A. During and Z. Takats, Analyst, 2012, 137, 4037–4044 RSC.
- J. Thunig, L. Flø, S. Pedersen-Bjergaard, S. H. Hansen and C. Janfelt, Rapid Commun. Mass Spectrom., 2012, 26, 133–140 CrossRef CAS.
- K. Nielsen, F. R. Lauritsen, T. Nissilä and R. A. Ketola, Rapid Commun. Mass Spectrom., 2012, 26, 297–303 CrossRef CAS.
- C. Janfelt and F. R. Lauritsen, Int. J. Mass Spectrom., 2011, 299, 178–183 CrossRef CAS.
- H. Wang, J. J. Liu, R. G. Cooks and Z. Ouyang, Angew. Chem., Int. Ed., 2010, 49, 877–880 CrossRef CAS.
- Z. Miao and H. Chen, J. Am. Soc. Mass Spectrom., 2009, 20, 10–19 CrossRef CAS.
- J. Thunig, S. H. Hansen and C. Janfelt, Anal. Chem., 2011, 83, 3256–3259 CrossRef CAS.
- D. R. Ifa, N. E. Manicke, A. L. Rusine and R. G. Cooks, Rapid Commun. Mass Spectrom., 2008, 22, 503–510 CrossRef CAS.
- A. El-Beqqali and M. Abdel-Rehim, J. Sep. Sci., 2007, 30, 2501–2505 CrossRef CAS.
- F. Versace, F. Sporkert, P. Mangin and C. Staub, Talanta, 2012, 101, 299–306 CrossRef CAS.
- M. Schulz and A. Schmoldt, Die Pharmazie - An International Journal of Pharmaceutical Sciences, 2003, 58, 447–474 CAS.
- N. Manicke, P. Abu-Rabie, N. Spooner, Z. Ouyang and R. G. Cooks, J. Am. Soc. Mass Spectrom., 2011, 22, 1501–1507 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |