 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fast and simultaneous analysis of biothiols by high-performance liquid chromatography with fluorescence detection under hydrophilic interaction chromatography conditions
Muneki
Isokawa
,
Takashi
Funatsu
and
Makoto
Tsunoda
*
Graduate School of Pharmaceutical Sciences, University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: makotot@mol.f.u-tokyo.ac.jp; Fax: +81-3-5802-3339
First published on 24th May 2013
Abstract
A method for analyzing biothiols based on high-performance liquid chromatography (HPLC)-fluorescence detection under hydrophilic interaction chromatography (HILIC) conditions has been developed. Thiols were derivatized with nonfluorescent ammonium 7-fluoro-2,1,3-benzoxadiazole-4-sulfonate (SBD-F), which selectively reacts with the thiol groups to furnish the corresponding fluorescent SBD–thiols. Among the six different kinds of HILIC columns examined, the ZIC-HILIC column with sulfobetaine groups in the stationary phase proved to be the best for the separation of SBD–thiols. Eight thiols—N-acetylcysteine, cysteamine, homocysteine, cysteine, cysteinylglycine, glutathione, γ-glutamylcysteine, and internal standard N-(2-mercaptopropionyl)glycine—were baseline-separated within 10 min. The detection sensitivity was improved partly due to the increase in the SBD–thiol fluorescence owing to the acetonitrile-rich mobile phase used. The detection limits at a signal-to-noise ratio of 3 were 0.02–3.4 nmol l−1. The method could successfully quantify six thiols in a human plasma sample, while cysteamine could not be detected. Both the intra- and interday precisions were below 4% for homocysteine, cysteine, cysteinylglycine, glutathione, and γ-glutamylcysteine except for N-acetylcysteine. This method should be a useful tool for investigating the relationship between sulfur metabolism and related diseases, since a multicomponent system consisting of different thiol compounds could be analyzed simultaneously with high sensitivity within a short time.
Introduction
Low-molecular-weight thiols such as cysteine (Cys), homocysteine (Hcy), N-acetylcysteine (NAC), glutathione (GSH), cysteinylglycine (CysGly), γ-glutamylcysteine (γ-GluCys), and cysteamine (CA) shown in Fig. 1A are metabolites of the sulfur cycle and play important roles in biological processes. Biothiols influence the extra- or intracellular redox state, help in eliminating free radicals, including reactive oxygen species, and remove toxic compounds.1 In addition, the sulfur metabolism is coupled with the one-carbon metabolism, where both DNA/RNA methylation and nucleotide/thymidylate synthesis take place.2 An imbalance in the sulfur metabolism causes many diseases such as vascular disease, Alzheimer's disease, human immunodeficiency virus infection, and cancer.3 Moreover, genetic defects in the enzymes regulating sulfur pools lead to homocystinuria, cystinuria, homocysteinemia, cysteinemia, and neural tube defects.3 Thus, simultaneous detection and quantification of different biothiols is important.4–9 Among the numerous analytical methods used for this purpose, HPLC-fluorescence detection has been the most widely used.9 Since thiols are nonfluorescent compounds, derivatization with a fluorescence reagent is needed. Many reagents containing maleimide and iodoacetamide groups have been used for the analysis of thiols.4–9 Halogenobenzofurazans such as 4-aminosulfonyl-7-fluoro-2,1,3-benzoxadiazole (ABD-F) and ammonium 7-fluoro-2,1,3-benzoxadiazole-4-sulfonate (SBD-F) also selectively react with thiols to furnish fluorescent derivatives, whereas the reagents and the by-products are nonfluorescent.10 SBD-F is more selective than ABD-F, and the reaction is shown in Fig. 1B. Most of the previous studies have used reversed-phase liquid chromatography (RPLC) for the separation of SBD–thiols.11–13 However, simultaneous detection of multiple thiols has not been successful because of the difficulty in retaining SBD–thiols owing to their high polarity.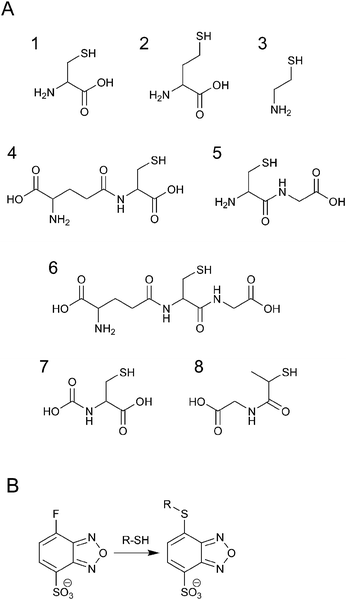 | ||
| Fig. 1 Chemical structures of SBD-F and thiols. (A) Structure of thiols: 1, Cys; 2, Hcy; 3, CA; 4, γ-GluCys; 5, CysGly; 6, GSH; 7, NAC; 8, MPG. (B) Reaction of SBD-F and thiols. | ||
Hydrophilic interaction chromatography (HILIC), which was first introduced in 1990 by A. J. Alpert,14 is advantageous for the retention and separation of polar, ionizable analytes.15–17 In HILIC, an organic-rich mobile phase, as in RPLC, and a polar stationary phase, as in normal-phase liquid chromatography, are used for the strong retention of polar compounds by hydrogen-bonding, ion exchange, and hydrophilic partitioning between the bulk mobile phase and the water-enriched layer on the polar stationary phase.14,16–24 Hence, we envisioned that HILIC could be useful for the simultaneous separation of different SBD–thiols. In addition, it was reported that the fluorescence intensity of SBD–thiols is higher in an acetonitrile-rich solution than in an aqueous solution.13 Hence, the use of an HILIC column would allow for the detection of SBD–thiols with better sensitivity. In this study, we aim to develop a method for the simultaneous analysis of different thiols derivatized with SBD-F reagent under HILIC conditions. Herein, we report that the eight thiols considered—N-(2-mercaptopropionyl)glycine (MPG), NAC, CA, Hcy, Cys, CysGly, GSH, and γ-GluCys—were well separated on the ZIC-HILIC column within 10 min. This validated method was applied successfully for the analysis of human plasma samples.
Experimental
Chemicals and reagents
L-Cysteine (Cys), DL-homocysteine (Hcy), L-glutathione (reduced form, GSH), cysteinylglycine (CysGly), and γ-glutamylcysteine (γ-GluCys) were purchased from Sigma-Aldrich (St. Louis, MO, USA). N-Acetyl-L-cysteine (NAC), tiopronin (N-(2-mercaptopropionyl) glycine, MPG), cysteamine (CA), trichloroacetic acid (TCA), and formic acid were obtained from Wako (Osaka, Japan). Ammonium 7-fluoro-2,1,3-benzoxadiazole-4-sulfonate (SBD-F) was purchased from Dojindo (Kumamoto, Japan). Tris(2-carboxyethyl)phosphine (TCEP) was obtained from TCI (Tokyo, Japan). Water was purified using a Milli-Q system (Millipore, Bedford, MA, USA). HPLC-grade acetonitrile was used. All other chemicals were of analytical-reagent grade.Preparation of standard and human plasma samples
Human plasma was purchased from Sigma-Aldrich. Sample preparation was performed based on a previous report, with little modification to the procedure.25 For the reduction of disulfides and protein-bound thiols, 5 μl of a 120 g l−1 TCEP solution in phosphate buffer was added to the mixture of 25 μl of a thiol aqueous solution or human plasma and 25 μl of a 3.4 μmol l−1 MPG aqueous solution as the internal standard. The resulting mixture was incubated for 30 min at room temperature. The sample was then mixed with 25 μl of a 100 g l−1 TCA solution and 1 mmol l−1 disodium ethylenediamine-N,N,N′,N′-tetraacetate (EDTA) solution. After centrifugation at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 10 min at 4 °C for deproteinization of plasma, 3 μl of 3.1 mol l−1 sodium hydroxide solution was added to 48 μl of the supernatant in order to neutralize the solution. A 50 μl aliquot of the mixture was added to the derivatization solution containing 125 μl of a 125 mmol l−1 borate buffer (pH 9.5), 4 mmol l−1 EDTA solution, and 50 μl of a 3.0 g l−1 SBD-F solution in the borate buffer. The resulting mixture was allowed to react for 60 min at 60 °C. The derivatization reaction was quenched by adding 25 μl of a 1 mol l−1 hydrochloric acid, and the resulting solution was cooled in ice. The injection samples contained 90% (v/v) acetonitrile for column selection and optimization of the mobile phase, and 75% (v/v) acetonitrile under the optimum conditions. Five microliters of the sample was injected into the HPLC system for analysis.
000 × g for 10 min at 4 °C for deproteinization of plasma, 3 μl of 3.1 mol l−1 sodium hydroxide solution was added to 48 μl of the supernatant in order to neutralize the solution. A 50 μl aliquot of the mixture was added to the derivatization solution containing 125 μl of a 125 mmol l−1 borate buffer (pH 9.5), 4 mmol l−1 EDTA solution, and 50 μl of a 3.0 g l−1 SBD-F solution in the borate buffer. The resulting mixture was allowed to react for 60 min at 60 °C. The derivatization reaction was quenched by adding 25 μl of a 1 mol l−1 hydrochloric acid, and the resulting solution was cooled in ice. The injection samples contained 90% (v/v) acetonitrile for column selection and optimization of the mobile phase, and 75% (v/v) acetonitrile under the optimum conditions. Five microliters of the sample was injected into the HPLC system for analysis.
HPLC apparatus and chromatographic conditions
The HPLC system consisted of a pump (PU-2080 Plus, JASCO, Tokyo, Japan), a column oven (860-CO, JASCO), and a fluorescence detector (RF-20A, Shimadzu, Kyoto, Japan). The column temperature was set at 35 °C, and the SBD–thiols were detected by fluorescence with excitation and emission wavelengths of 375 and 510 nm, respectively. The chromatograms were analyzed using the software Chromato-Pro (Run Time Corporation, Kanagawa, Japan). In order to select a suitable column for thiol separation, a mixture of eight SBD–thiols was injected into six HILIC columns (ZIC-HILIC (150 mm × 2.1 mm i.d., 5 μm, Merck, Germany), Inertsil Amide (150 mm × 3.0 mm i.d., 5 μm, GL Sciences, Tokyo, Japan), Inertsil Diol (150 mm × 4.6 mm i.d., 5 μm, GL Sciences), Inertsil SIL (150 mm × 3.0 mm i.d., 5 μm, GL Sciences), PC HILIC (150 mm × 2.0 mm i.d., 5 μm, Shiseido, Tokyo, Japan), and TSKgel NH2-100 (150 mm × 2.0 mm i.d., 3 μm, Tosoh, Tokyo, Japan)). Acetonitrile–10 mmol l−1 ammonium formate buffer (pH 3.0) (75/25, v/v) was used for the mobile phase. The linear velocity of the mobile phase was 58 mm min−1.Optimization for the separation of SBD–thiols was carried out on a ZIC-HILIC column. The initial mobile phase was acetonitrile–10 mmol l−1 ammonium formate buffer (pH 3.0) (75/25, v/v) and the flow rate was 0.2 ml min−1. The acetonitrile content, buffer pH, and concentration of salt in the buffer were optimized first, and then, the flow rate and acetonitrile content in the injection sample were determined.
Calculation of the log D value
SPARC software26 was used to calculate the log D values of the SBD–thiols. Log D is defined as the logarithm of the ratio of equilibrium concentrations of a species in both neutral and ionized forms of a certain molecule in an organic solvent to the concentration of the same species in the water phase. Calculations were performed under the following conditions: organic solvent, acetonitrile; ionic strength of the water phase, 0; and temperature, 25 °C.Measurement of pH and apparent pH 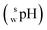
The 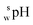 value of a substance is measured in a mixture of aqueous and organic solutions, while its pH is measured in an aqueous solution. A pH meter calibrated in aqueous buffers was used for the measurement of both pH and
value of a substance is measured in a mixture of aqueous and organic solutions, while its pH is measured in an aqueous solution. A pH meter calibrated in aqueous buffers was used for the measurement of both pH and 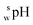 .
.
Fluorometry
SBD–thiol aqueous solutions were obtained by the derivatization method mentioned above. In order to examine the effect of the dissolving solution on the fluorescence intensity of the SBD–thiols, the test solution was diluted with water or acetonitrile. The final concentration of the SBD–thiol solution was set at 20 μmol l−1, and the solution contained 0 or 75% (v/v) acetonitrile. The change in the fluorescence intensity of 20 μmol l−1 SBD–Cys dissolved in a solution containing 10 to 90% (v/v) acetonitrile was also investigated. The fluorescence spectra of the thiols were measured using a FP-6500 (JASCO) spectrofluorometer.Validation
Five standard concentrations of the thiols covering the range of 1–100 nmol l−1 for NAC, 0.5–50 nmol l−1 for CA, 15–1500 nmol l−1 for Hcy and γ-GluCys, 60–6000 nmol l−1 for Cys, 7.5–750 nmol l−1 for CysGly, and 10–1000 nmol l−1 for GSH were injected as calibration samples. Recovery of the thiols in plasma was assessed by spiking additional thiols at three different concentrations. The intra- and interday precisions were assessed by replicate analysis of the same plasma sample on the same day and on sequential days, respectively. In the recovery and reproducibility analyses, the samples were injected six times at each concentration and then, the relative peak height to MPG (internal standard) was evaluated.Results and discussion
Column selection
There are many kinds of HILIC columns available, and the functional groups on their support can affect the separation efficiency.17–22 Thus, six kinds of columns (Inertsil SIL, Inertsil Amide, Inertsil Diol, TSKgel NH2-100, PC HILIC, and ZIC-HILIC, Table 1) were investigated in order to identify the most suitable column for the separation of SBD–thiols, using an acetonitrile–10 mmol l−1 ammonium formate buffer (pH 3.0) (75/25, v/v) as the mobile phase.
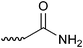
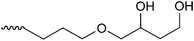

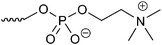
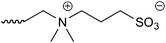
Fig. 2 shows the retention time of each SBD–thiol with the HILIC column investigated. TSKgel NH2-100 was excluded because the SBD–thiols were so strongly retained on TSKgel NH2-100 that the last peak in the elution profile appeared after 6 h. This was probably due to the strong anion-exchange property of the amino group in the stationary phase of the column. SBD–thiols may be anionic because of the presence of a sulfate group.
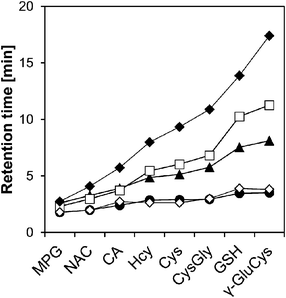 | ||
| Fig. 2 Retention time of SBD–thiols on five HILIC columns. Columns: Inertsil Diol, ●; Inertsil SIL, ◇; PC HILIC, ▲; Inertsil Amide, □; ZIC-HILIC, ◆. Column temperature: 35 °C. Mobile phase: acetonitrile–10 mmol l−1 ammonium formate buffer (pH 3.0) = 75/25 (v/v). Linear velocity: 58 mm min−1. Fluorescence detection: ex; 375 nm, em; 510 nm. | ||
The elution order of thiol derivatives after the run was almost the same for the five columns, which indicated that the main retention mechanism is similar for all the HILIC columns studied, despite the difference in their functional groups. As shown in Fig. 2, the SBD–thiols were strongly retained on Inertsil Amide, PC HILIC, and ZIC-HILIC but weakly retained on Inertsil SIL and Inertsil Diol. This result agreed with the characterization of HILIC columns in previous reports: amide and zwitterionic columns are strongly hydrophilic, while diol and bare silica columns are weakly hydrophilic.19 Weak retention of thiols on Inertsil Diol and Inertsil SIL resulted in the co-elution of SBD–Hcy and SBD–Cys. A resolution of less than 1.5 was observed for SBD–Hcy and SBD–Cys on PC HILIC, as well as in the case of SBD–GSH and SBD–γ-GluCys on Inertsil Amide. Accordingly, ZIC-HILIC was selected for further studies.
Optimization of the mobile phase for the separation of SBD–thiols on the ZIC-HILIC column
Previous reports proposed that the retention on HILIC columns is mainly caused by partitioning of the analyte between the relatively hydrophobic mobile phase and the water-enriched layer immobilized on the stationary phase, and partly by hydrogen-bonding and electrostatic interactions.14,16–23 Thus, in order to find the optimum constituents of the mobile phase for the separation of SBD–thiols under HILIC conditions, the ratio of acetonitrile to water (for the former mechanism), pH, and salt concentration of the buffer (for the latter mechanism) in the mobile phase were examined.The primary mobile phase was acetonitrile–10 mmol l−1 ammonium formate buffer (pH 3.0) (75/25, v/v).18 First, the effect of acetonitrile content (70%, 75%, and 80%) in the mobile phase was examined. As shown in Fig. 3A, SBD–thiols were strongly retained with an increase in the acetonitrile content. The elution followed the order MPG, NAC, CA, Hcy, Cys, CysGly, GSH, and γ-GluCys under all the conditions examined. However, this order was not exactly opposite to that observed in RPLC, where the elution order was Cys, Hcy, CysGly, GSH, and MPG with a mobile phase of 0.1 mol l−1 acetate buffer (pH 4.5) in methanol.11 This result indicated that the separation mechanism under HILIC conditions does not include only partitioning, the dominant separation mechanism for RPLC mode. Further, the peaks were not well separated at 70% acetonitrile, while SBD–GSH and SBD–γ-GluCys were strongly retained at 80% acetonitrile (35 and 44 min, respectively). Thus, 75% (v/v) acetonitrile was selected as the optimum ratio.
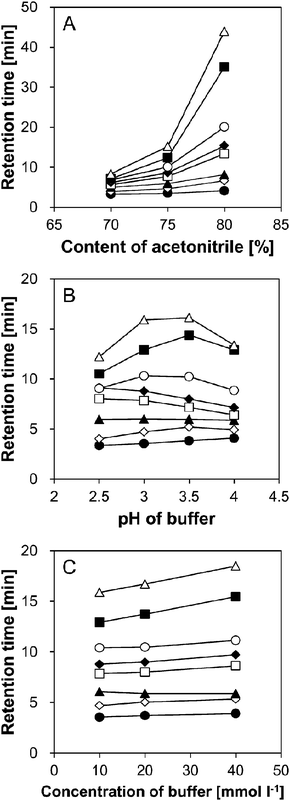 | ||
| Fig. 3 Effect of (A) acetonitrile content, (B) pH of ammonium formate buffer and (C) concentration of ammonium formate buffer on retention time of SBD–thiols. Symbols: SBD–MPG, ●; SBD–NAC,◇; SBD–CA, ▲; SBD–Hcy, □; SBD–Cys, ◆; SBD–CysGly, ○; SBD–GSH, ■; SBD–γ-GluCys, △. Column: ZIC-HILIC (150 mm × 2.1 mm i.d., 5 μm, Merck). Column temperature: 35 °C. Mobile phase: (A) acetonitrile–10 mmol l−1 ammonium formate buffer (pH 3.0), (B) acetonitrile–10 mmol l−1 ammonium formate buffer = 75/25 (v/v), and (C) acetonitrile–ammonium formate buffer (pH 3.0) = 75/25. Flow rate: 0.2 ml min−1. Fluorescence detection: ex 375 nm, em 510 nm. Injection sample: 5 μl, containing 90% acetonitrile. | ||
Next, the effect of buffer pH on the retention of SBD–thiols was examined. Though the pH of the mobile phase did not affect the charge state of the sulfate and quaternary amino groups in the stationary phase, the state of SBD–thiols having an amino or a carboxyl group could be affected by the environmental pH in the mobile phase. As shown in Fig. 3B, the effects of pH were not the same for all the SBD–thiols. While SBD–Cys and SBD–Hcy eluted slightly faster with the increase in pH, SBD–γ-GluCys and SBD–GSH were retained most strongly at pH 3.5. In order to investigate the relationship between the retention behavior and the pH of the mobile phase, log D values, i.e., the acetonitrile–water distribution coefficients, of SBD–thiols, were calculated. Log D value is an index of hydrophilicity of a molecule, and reflects the charge state of the molecule. According to the calculations, SBD–γ-GluCys should become more hydrophilic with an increase in pH from 1.5 to 5.5, with the most hydrophilic behavior at pH 5.5. This result confirmed the retention behavior shown in Fig. 3B, when the apparent pH 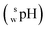 16 in the acetonitrile–water mixture was considered. The
16 in the acetonitrile–water mixture was considered. The 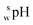 values of acetonitrile–10 mmol l−1 ammonium formate buffer (75/25, v/v) at pH 2.5, 3.0, 3.5, and 4.0 were around 4.0, 4.6, 5.1, and 5.6, respectively. Considering the shift in pH, the retention behavior of other SBD–thiols agrees well with the change in log D. This result indicated that hydrophilic partitioning might be the main retention mechanism for the retention of SBD–thiols rather than electrostatic interactions. Further, SBD–Cys and SBD–CysGly were not separated at pH 2.5. Since resolution was lower at pH 3.5 and 4.0 than at pH 3.0, the latter pH value was considered optimal for the mobile phase.
values of acetonitrile–10 mmol l−1 ammonium formate buffer (75/25, v/v) at pH 2.5, 3.0, 3.5, and 4.0 were around 4.0, 4.6, 5.1, and 5.6, respectively. Considering the shift in pH, the retention behavior of other SBD–thiols agrees well with the change in log D. This result indicated that hydrophilic partitioning might be the main retention mechanism for the retention of SBD–thiols rather than electrostatic interactions. Further, SBD–Cys and SBD–CysGly were not separated at pH 2.5. Since resolution was lower at pH 3.5 and 4.0 than at pH 3.0, the latter pH value was considered optimal for the mobile phase.
Finally, the effect of buffer concentration on the mobile phase was investigated. Higher buffer concentrations caused retention of the SBD–thiols for a longer time, especially SBD–GSH and SBD–γ-GluCys (Fig. 3C). There may be two reasons for this behavior: (1) at a higher buffer concentration, electrostatic repulsion between the sulfate groups in the stationary phase and the SBD–thiols is suppressed, and (2) the water-enriched layer on the stationary phase, which also works as a pseudo-stationary phase under the HILIC conditions, is thickened at higher buffer concentrations.27–30 Since higher buffer concentrations resulted in longer analysis time and buffer concentrations lower than 10 mmol l−1 resulted in low buffering ability, 10 mmol l−1 was chosen as the optimal concentration of the ammonium formate buffer. Consequently, an acetonitrile solution containing 25% (v/v) 10 mmol l−1 ammonium formate buffer (pH 3.0) was selected as the optimal constituent for the mobile phase in the analysis of SBD–thiols using the ZIC-HILIC column.
The water content in the injection samples could affect the peak shape in HILIC.31 Though the water content of the aqueous solution in the injection sample was raised from 10% to 35%, there was no significant effect on the peak shape. While the number of theoretical plates of the SBD–thiols slightly decreased with the increase in the water content in the sample, the effect was not significant (less than 10% decrease). Thus, the water content in the injection sample was kept at 25% (v/v), as in the mobile phase.
The reduced viscosity resulting from the acetonitrile-rich mobile phase used in HILIC could allow for analysis at higher flow rates as compared to those in RPLC.32 Thus, for faster separation, the flow rate was increased from 0.2 to 0.4 ml min−1, resulting in an acceptable back pressure (<5 MPa). Fig. 4A shows a typical chromatogram of the standards of the SBD–thiols under the optimized conditions. Baseline separation was achieved within 10 min, and all the peaks of the SBD–thiols were sharp and symmetrical.
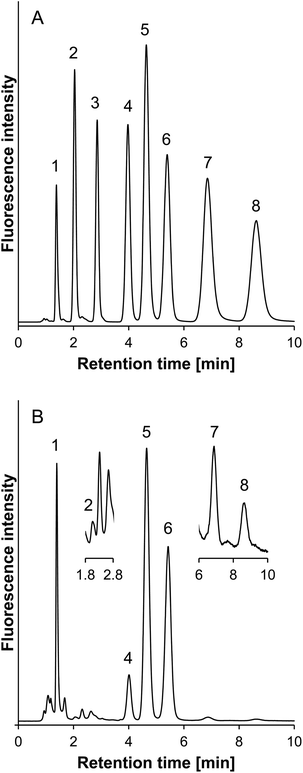 | ||
| Fig. 4 Typical chromatogram of (A) the standard sample and (B) the human plasma sample. Peaks; 1, SBD–MPG; 2, SBD–NAC; 3, SBD–CA; 4, SBD–Hcy; 5, SBD–Cys; 6, SBD–CysGly; 7, SBD–GSH; 8, SBD–γ-GluCys. Column: ZIC-HILIC (150 mm × 2.1 mm i.d., 5 μm, Merck). Column temperature: 35 °C. Mobile phase: acetonitrile–10 mmol l−1 ammonium formate buffer (pH 3.0) = 75/25 (v/v). Flow rate: 0.4 ml min−1. Fluorescence detection: ex 375 nm, em 510 nm. Injection sample: 5 μl, containing 75% acetonitrile. | ||
Analysis of thiols in the human plasma sample
Separation of thiols in the human plasma sample was performed under the optimal conditions. As shown in Fig. 4B, peaks of SBD–thiols except SBD–CA were found without any interference in the chromatogram. The concentrations of thiols in human plasma were determined as follows: 0.15 μmol l−1 for NAC, 8.7 μmol l−1 for Hcy, 197.1 μmol l−1 for Cys, 19.9 μmol l−1 for CysGly, 0.94 μmol l−1 for GSH, and 1.1 μmol l−1 for γ-GluCys. These values were in good agreement with those reported in the previous studies.11,12,33–36Fluorescence intensity of SBD–thiols
As mentioned earlier, it has been reported that the fluorescence intensity of SBD–thiols varied with the change in the solvent system.13 Thus, the effect of acetonitrile content in the solvent on the fluorescence intensity of SBD–thiols was investigated. An increase in the fluorescence intensity of SBD–Cys was observed with an increase in the acetonitrile content (data not shown). The relative fluorescence intensity of SBD–thiols in a mixture of acetonitrile and water (75/25, v/v) to that in water was determined to be as follows: 2.4 for SBD–MPG, 3.1 for SBD–NAC, 1.2 for SBD–CA, 2.8 for SBD–Hcy, 3.9 for SBD–Cys, 1.0 for SBD–CysGly, 1.7 for SBD–GSH, and 3.8 for SBD–γ-GluCys. This indicated that the detection of SBD–thiols should be more sensitive under HILIC conditions with an acetonitrile-rich mobile phase than under RPLC conditions with a water-rich mobile phase. As mentioned below, the sensitivity of SBD–thiols under HILIC conditions was actually higher than that under RPLC conditions.Validation of the developed method
Table 2 shows the linear range, the limit of detection when the signal-to-noise (S/N) ratio is 3 and the limit of quantitation when the S/N ratio is 10. As shown in Table 3, the developed method was faster and more sensitive than those using halogenobenzofurazans12,34 and other fluorescence reagents, which were presented in previous reports.35–39 The improvement of sensitivity with SBD-F under HILIC conditions was partly due to the increase in the fluorescence intensity of the derivatized thiols in the organic-rich mobile phase, as described above. Table 3 also shows that the proposed method would be suitable for the fast and simultaneous analysis of biothiols, including γ-GluCys.| LOD [nmol l−1] S/N = 3 | LOQ [nmol l−1] S/N = 10 | Linearity [nmol l−1] r2 > 0.999 | |
|---|---|---|---|
| NAC | 0.3 | 0.8 | 1–100 |
| CA | 0.02 | 0.07 | 0.5–50 |
| Hcy | 0.8 | 2.6 | 15–1500 |
| Cys | 1.5 | 5.0 | 60–6000 |
| CysGly | 0.3 | 0.9 | 7.5–750 |
| GSH | 0.8 | 2.7 | 10–1000 |
| γ-GluCys | 3.4 | 12 | 15–1500 |
| Labeling reagent | LOD [nmol l−1] (S/N = 3) | Separation mode | Analysis time [min] | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| NAC | CA | Hcy | Cys | CysGly | GSH | γ-GluCys | ||||
| a Abbreviations: SBD-BF, ammonium 5-bromo-7-fluorobenzo-2-oxa-1,3-diazole-4-sulphonate; IAB, 3-iodoacetylaminobenzanthrone; TMPAB-I, 1,3,5,7-tetramethyl-8-phenyl-(4-iodoacetamido)difluoroboradiaza-s-indacene; MIAC, N-(2-acridonyl)maleimide; and mBrB, bromobimane. b There were some unknown peaks. c S/N ratio was 5. d There were some split peaks. | ||||||||||
| SBD-F | 0.3 | 0.02 | 0.8 | 1.5 | 0.3 | 0.8 | 3.4 | HILIC-isocratic | 10 | This study |
| SBD-Fb | 30 | 160 | 470 | RPLC-isocratic | 10 | 12 | ||||
| SBD-BFc | 10 | 100 | 10 | 20 | RPLC-gradient | 12 | 34 | |||
| IAB | 2 | 2.3 | 1 | 1 | RPLC-isocratic | 55 | 37 | |||
| TMPAB-I | 0.3 | 0.7 | 0.3 | 0.3 | RPLC-isocratic | 20 | 36 | |||
| MIACb | 60 | 70 | 100 | RPLC-stepwise | 6 | 35 | ||||
| N-(2,4-Dinitrophenyl-aminoethyl)maleimided | 500 | 500 | 500 | 500 | 500 | RPLC-gradient | 45 | 38 | ||
| mBrB | 50 | 50 | 50 | 50 | 50 | RPLC-gradient | 6 | 39 | ||
The recoveries of all SBD–thiols in the plasma sample were in the range 94–109%. The intraday precision was below 4% as relative standard deviation. While the interday precision for NAC was 8.94%, those for the other thiols were lower than 4%. Table 4 shows the recovery and intraday precision in detail.
| Thiols (n = 6) (added/μmol l−1) | Concentration (mean ± SD/μmol l−1) | Precision (R.S.D., %) | Recovery (%) |
|---|---|---|---|
| NAC | |||
| 0 | 0.15 ± 0.004 | 2.37 | — |
| 0.3 | 0.47 ± 0.006 | 1.00 | 105 |
| 0.6 | 0.76 ± 0.004 | 0.46 | 102 |
| 1.2 | 1.43 ± 0.005 | 0.35 | 106 |
| Hcy | |||
| 0 | 8.7 ± 0.06 | 0.73 | — |
| 3.0 | 12.0 ± 0.08 | 0.67 | 103 |
| 6.0 | 14.9 ± 0.05 | 0.32 | 101 |
| 12.0 | 22.6 ± 0.07 | 0.33 | 109 |
| Cys | |||
| 0 | 196.8 ± 1.4 | 0.72 | — |
| 60 | 251.0 ± 2.3 | 0.90 | 98 |
| 120 | 298.5 ± 1.1 | 0.36 | 94 |
| 240 | 437.2 ± 1.3 | 0.31 | 100 |
| CysGly | |||
| 0 | 19.9 ± 0.06 | 0.33 | — |
| 9 | 28.8 ± 0.10 | 0.35 | 100 |
| 18 | 37.0 ± 0.08 | 0.23 | 98 |
| 36 | 58.7 ± 0.15 | 0.25 | 105 |
| GSH | |||
| 0 | 0.94 ± 0.02 | 1.93 | — |
| 1.5 | 2.42 ± 0.03 | 1.29 | 99 |
| 3.0 | 3.85 ± 0.01 | 0.23 | 98 |
| 6.0 | 7.07 ± 0.04 | 0.51 | 102 |
| γ-GluCys | |||
| 0 | 1.14 ± 0.02 | 2.16 | — |
| 1.5 | 2.54 ± 0.10 | 3.91 | 96 |
| 3.0 | 3.89 ± 0.03 | 0.84 | 94 |
| 6.0 | 7.03 ± 0.03 | 0.50 | 98 |
Conclusions
Biothiols derivatized with SBD-F were successfully analyzed under the optimal HILIC conditions. To the best of our knowledge, this is the first attempt to analyze the low-molecular-weight thiols under HILIC conditions, and to utilize fluorescence property of SBD–thiols for sensitive detection. The developed method allowed for simultaneous, fast, and highly sensitive analysis of biothiols—NAC, Hcy, Cys, CysGly, GSH, and γ-GluCys—with an isocratic mobile phase. The high sensitivity probably resulted from the stronger fluorescence of SBD–thiols in the more organic-rich solvent. The proposed method should be useful in investigating the relationship between sulfur metabolism, in particular γ-GluCys, and related diseases. This report would also be helpful to develop a method for analyzing zwitterionic compounds under HILIC conditions.Acknowledgements
The authors acknowledge their sincere gratitude to GL Sciences, Merck, Shiseido, and Tosoh for providing the Inertsil series (Inertsil SIL, Inertsil Diol, and Inertsil Amide), ZIC-HILIC, PC HILIC, and TSKgel NH2-100 columns, respectively.References
- N. J. Pace and E. Weerapana, ACS Chem. Biol., 2013, 8, 283 CrossRef CAS.
- S. W. Choi and J. B. Mason, J. Nutr., 2000, 130, 129 CAS.
- D. M. Townsend, K. D. Tew and H. Tapiero, Biomed. Pharmacother., 2004, 58, 47 CrossRef CAS.
- K. Shimada and K. Mitamura, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 1994, 659, 227 CrossRef CAS.
- T. Toyo'oka, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2009, 877, 3318 CrossRef CAS.
- K. Kuśmierek, G. Chwatko, R. Głowacki and E. Bald, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2009, 877, 3300 CrossRef.
- K. Kuśmierek, G. Chwatko, R. Głowacki, P. Kubalczyk and E. Bald, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2011, 879, 1290 CrossRef.
- R. E. Hansen and J. R. Winther, Anal. Biochem., 2009, 394, 147 CrossRef CAS.
- M. E. McMenamin, J. Himmelfarb and T. D. Nolin, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2009, 877, 3274 CrossRef CAS.
- S. Uchiyama, T. Santa, N. Okiyama, T. Fukushima and K. Imai, Biomed. Chromatogr., 2001, 15, 295 CrossRef CAS.
- T. D. Nolin, M. E. McMenamin and J. Himmelfarb, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2007, 852, 554 CrossRef CAS.
- S. Ichinose, M. Nakamura, M. Maeda, R. Ikeda, M. Wada, M. Nakazato, Y. Ohba, N. Takamura, T. Maeda, K. Aoyagi and K. Nakashima, Biomed. Chromatogr., 2009, 23, 935 CrossRef CAS.
- T. Toyo'oka and K. Imai, Analyst, 1984, 109, 1003 RSC.
- A. J. Alpert, J. Chromatogr., 1990, 499, 177 CrossRef CAS.
- M. Lämmerhofer, J. Sep. Sci., 2010, 33, 679 CrossRef.
- D. V. McCalley, J. Chromatogr., A, 2007, 1171, 46 CrossRef CAS.
- B. Buszewski and S. Noga, Anal. Bioanal. Chem., 2011, 402, 231 CrossRef.
- R.-I. Chirita, C. West, A.-L. Finaru and C. Elfakir, J. Chromatogr., A, 2010, 1217, 3091 CrossRef CAS.
- Y. Kawachi, T. Ikegami, H. Takubo, Y. Ikegami, M. Miyamoto and N. Tanaka, J. Chromatogr., A, 2011, 1218, 5903 CrossRef CAS.
- Y. Guo and S. Gaiki, J. Chromatogr., A, 2011, 1218, 5920 CrossRef CAS.
- N. P. Dinh, T. Jonsson and K. Irgum, J. Chromatogr., A, 2011, 1218, 5880 CrossRef CAS.
- A. Periat, B. Debrus, S. Rudaz and D. Guillarme, J. Chromatogr., A, 2013, 1282, 72 CrossRef CAS.
- W. Bicker, J. Wu, M. Lämmerhofer and W. Lindner, J. Sep. Sci., 2008, 31, 2971 CrossRef CAS.
- T. Zhang, D. J. Creek, M. P. Barrett, G. Blackburn and D. G. Watson, Anal. Chem., 2012, 84, 1994 CrossRef CAS.
- J. Krijt, M. Vackova and V. Kozich, Clin. Chem., 2001, 47, 1821 CAS.
- SPARC release w4.6.1691-s4.6.1687, http://ibmlc2.chem.uga.edu/sparc/, 2013.
- Y. Guo and S. Gaiki, J. Chromatogr., A, 2005, 1074, 71 CrossRef CAS.
- R.-I. Chirita, C. West, S. Zubrzycki, A.-L. Finaru and C. Elfakir, J. Chromatogr., A, 2011, 1218, 5939 CrossRef CAS.
- D. V. McCalley, J. Chromatogr., A, 2010, 1217, 3408 CrossRef CAS.
- G. Greco, S. Grosse and T. Letzel, J. Chromatogr., A, 2012, 1235, 60 CrossRef CAS.
- J. Ruta, S. Rudaz, D. V. McCalley, J. L. Veuthey and D. Guillarme, J. Chromatogr., A, 2010, 1217, 8230 CrossRef CAS.
- B. Chauve, D. Guillarme, P. Cléon and J.-L. Veuthey, J. Sep. Sci., 2010, 33, 752 CrossRef CAS.
- A. Andersson, A. Isaksson, L. Brattstrom and B. Hultberg, Clin. Chem., 1993, 39, 1590 CAS.
- G. Cevasco, A. M. Piątek, C. Scapolla and S. Thea, J. Chromatogr., A, 2010, 1217, 2158 CrossRef CAS.
- B. Benkova, V. Lozanov, I. P. Ivanov, A. Todorova, I. Milanov and V. Mitev, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2008, 870, 103 CrossRef CAS.
- X.-F. Guo, H. Wang, Y.-H. Guo, Z.-X. Zhang and H.-S. Zhang, J. Chromatogr., A, 2009, 1216, 3874 CrossRef CAS.
- H. Wang, S.-C. Liang, Z.-M. Zhang and H.-S. Zhang, Anal. Chim. Acta, 2004, 512, 281 CrossRef CAS.
- S. Ohmori, T. Kawase, M. Higashiura, Y. Chisaka, K. Nakata and Y. Yamasaki, J. Chromatogr., B: Biomed. Sci. Appl., 2001, 762, 25 CrossRef CAS.
- A. Pastore, R. Massoud, C. Motti, A. Lo Russo, G. Fucci, C. Cortese and G. Federici, Clin. Chem., 1998, 44, 825 CAS.
| This journal is © The Royal Society of Chemistry 2013 |