 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynchrotron FTIR reveals lipid around and within amyloid plaques in transgenic mice and Alzheimer's disease brain
Catherine R.
Liao
*a,
Margaret
Rak
*a,
Jillian
Lund
a,
Miriam
Unger
b,
Eric
Platt
c,
Benedict C.
Albensi
c,
Carol J.
Hirschmugl
d and
Kathleen M.
Gough
a
aDepartment of Chemistry, University of Manitoba, 360 Parker Building,144 Dysart Road, Winnipeg, Manitoba, Canada. E-mail: Kathleen.Gough@ad.umanitoba.ca; Fax: +1-204-474-7608; Tel: +1-204-474-6262
bSynchrotron Radiation Center, University of Wisconsin-Madison, Stoughton, WI 53589, USA
cDepartment of Pharmacology and Therapeutics, Faculty of Medicine, University of Manitoba and Division of Neurodegenerative Disorders, St. Boniface Hospital Research Centre, Winnipeg, Manitoba R3E 0W3, Canada. E-mail: BAlbensi@sbrc.ca; Fax: +1-204-237-4092; Tel: +1-204-235-3942
dDepartment of Physics, University of Wisconsin-Milwaukee, Milwaukee, WI 53211, USA. E-mail: cjhirsch@uwm.edu; Fax: +1-414.229.5589; Tel: +1-414-229 5748
First published on 28th March 2013
Abstract
While the basis of neuronal degeneration in Alzheimer's disease (AD) continues to be debated, the amyloid cascade hypothesis remains central. Amyloid plaques are a required pathological marker for post mortem diagnosis, and Aβ peptide is regarded by most as a critical trigger at the very least. We present spectrochemical image analysis of brain tissue sections obtained with the mid-infrared beamline IRENI (InfraRed ENvironmental Imaging, Synchrotron Radiation Center, U Wisconsin-Madison), where the pixel resolution of 0.54 × 0.54 µm2 permits analysis at sub-cellular dimensions. Spectrochemical images of dense core plaque found in hippocampus and cortex sections of two transgenic mouse models of AD (TgCRND8 and 3×Tg) are compared with plaque images from a 91 year old apoE43 human AD case. Spectral analysis was done in conjunction with histochemical stains of serial sections. A lipid membrane-like spectral signature surrounded and infiltrated the dense core plaques in all cases. Remarkable compositional similarities in early stage plaques suggest similar routes to plaque formation, regardless of genetic predisposition or mammalian origin.
Introduction
Alzheimer's disease (AD) is a complex neurodegenerative disorder that manifests clinically as progressive memory loss, and is diagnosed through post mortem analyses of brain tissue that reveal the classical lesions known as senile plaques (SP) and neurofibrillary tangles (NFT).1 The former consist of β-sheet aggregated peptides (Aβ) from mis-processing of the amyloid protein precursor (APP), while the NFT are formed from tau proteins, a microtubule stabilizer that forms paired helical filaments upon hyperphosphorylation. Alzheimer's disease presents a major and growing health concern worldwide since the number of AD patients steadily increases as the average population ages.2The most recent guidelines for diagnostic criteria for AD3–6 affirm the requirement for the presence of at least a moderate number of neuritic Aβ plaques that may be observed in stained sections of post mortem neocortical tissue as well as the regional distribution of NFT at or above Braak stage IV. The latter show a stronger correlation with clinical tests for cognitive impairment.7–9 Despite several decades of effort, the few approved therapies provide only some small symptomatic relief; efforts to halt, reverse or prevent AD are so far unsuccessful.10 Research into prevention and cure has focussed mainly on BACE inhibitors and anti-Aβ vaccines, since the plaques, and also the protofibrils and oligomers of Aβ, are presumed to be the critical players in disease evolution. Hypotheses concerning triggers and progression continue to change. The correlation between NFT and cognitive impairment, for example, is taken by some to mean that the Aβ is a necessary trigger, but not necessarily the continued driving force behind the disease and that NFT, not Aβ, represents the more critical target. However, the report on diagnostic criteria3 underlines the fact that the genetic factors associated with early onset familial AD (FAD) are all associated with Aβ. The only consistent genetic risk factor for late onset AD (LOAD) is the APOE ε4 allele, responsible for the ApoE4 apolipoprotein, which also is reported to interact with Aβ.11
We have been using vibrational spectrochemical imaging to study unfixed, snap-frozen, dessicated tissue sections from human AD brain and from transgenic mice, in an effort to identify cellular and subcellular features that might be overlooked or altered in histopathological and immunohistochemical methods.12–16 Two major findings have been the discovery of elevated quantities of creatine crystals in transgenic mouse brain13,15 and the appearance of a lipid halo around dense core, but not diffuse, Aβ plaques.14–16
Since the transgenic mouse models are developed from FAD mutations, we have now begun to compare them to plaque from LOAD. Brain tissue was acquired from two transgenic mouse models of AD. TgCRND8 mice express the Swedish K670N/M671L and Indiana V717F FAD mutations in the human amyloid precursor protein (APP).17 Triply transgenic 3×Tg mice express the KM670/671NL mutation in APP, the presenilin mutation PS1 (M146V), and human four-repeat Tau harbouring the P301L mutation.18 Dense core plaques manifest by 3–5 months in the TgCRND8 mice17 but not until ∼1 year in the 3×Tg.18 In order to compare plaques in the early stages of formation across the two animal models, this study included two 5 month and one 8 month TgCRND8 animals, and one 12 month, four 15 month, and three 17 month 3×Tg animals. Autopsy tissue from a 91 year old human apoE43 case was imaged for comparison. All FTIR spectrochemical images were obtained at the IRENI beam line (Synchrotron Radiation Center, WI). At IRENI, a 320 mrad swath of light is extracted, converted into 12 beams and reorganized into a 3 × 4 matrix to illuminate a Focal Plane Array detector (Bruker Hyperion 3000 FTIR microscope). FTIR imaging with IRENI enables the highest possible IR spatial resolution. We present here the first results into the composition of plaque from this on-going investigation.
Methods
Mouse tissue preparation
The details of sample acquisition and preparation are as described in Kastyak et al.16 All experimental protocols for animal studies were approved by appointed Protocol Management Review committees at the University of Toronto and University of Manitoba, following guidelines established by the Canadian Council for Animal Care. The frozen, unfixed tissues were cryotomed at 8 µm thickness and mounted on BaF2 windows. The samples were stored frozen until data collection to minimize oxidative degradation of sensitive functional groups, particularly unsaturated bonds, such as![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) C
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C
C![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) and CO.19 Serial sections were placed on glass slides and stained with Congo red (Sigma-Aldrich), counterstained with Mayer's hematoxylin (Sigma-Aldrich), dehydrated and mounted with Permount (Fisher).
and CO.19 Serial sections were placed on glass slides and stained with Congo red (Sigma-Aldrich), counterstained with Mayer's hematoxylin (Sigma-Aldrich), dehydrated and mounted with Permount (Fisher).
Human autopsy AD tissues
Unfixed, frozen hippocampal tissue came from a 91 year old female patient with advanced AD (Braak 6 (ref. 20)). The tissue was obtained from the Netherlands Brain Bank (NBB), Netherlands Institute for Neuroscience, Amsterdam. All material has been collected from donors for or from whom a written informed consent for a brain autopsy and the use of the material and clinical information for research purposes had been obtained by the NBB. The sample was prepared as described above. A total of 5 hippocampal dense-cored plaques were imaged at the IRENI beamline. Their identity was confirmed with Congo red staining.FTIR data collection
Synchrotron FTIR (sFTIR) image data were recorded in transmission mode at the IRENI beamline (SRC, University of Wisconsin-Madison). sFTIR images were acquired using a 74× objective (Ealing Inc., Rocklin, CA, USA), yielding a pixel size of 0.54 × 0.54 µm2 on the Focal Plane Array (FPA).21,22 Between 64 and 256 scans were co-added in order to obtain good signal to noise. The regions analyzed for this study included dense-cored plaques and the surrounding tissue. Each tile was selected to be 64 × 64 pixels; mosaic images are comprised of 3 × 3, to 3 × 1 tiles.FTIR spectral analysis
Spectrochemical images based on the integrated intensity of relevant peaks were created with OPUS software (Version 6.0, Bruker) from original, unprocessed spectra. The lipid distribution was based on the area of the symmetric CH2 band at 2850 cm−1 (2858–2848 cm−1, baseline 3004–2760 cm−1). Dense core plaque was imaged from the area of the β-sheet amide I shoulder at 1630 cm−1 (1640–1620 cm−1, baseline 1806–1485 cm−1) or the amyloid band at 1390 cm−1 (1415–1370 cm−1, same baseline).16 All sections were cut at the same thickness; all processed results are displayed on the same false-colour scale: red = high through to blue = low or absent, for the given feature.The geometric pixel size is fixed for the given optics, while the spatial resolution is diffraction limited, with a trend line of about 0.47 × λ for this system. This means that all wavelengths are oversampled, throughout the entire spectrum. Actual spatial resolution in the CH stretch wavelength range is on the order of 1–2 µm, while that for the amide I region is about 3 µm.
Results
Our earlier studies have demonstrated that dense core plaques in the TgCRND8 mouse brain are always enveloped by elevated lipid, in brains from animals aged 5 to 21 months.14 More recently, images of plaque in a 15 month old TgCRND8 mouse brain, collected at higher spatial resolution with IRENI, demonstrated that the lipid signature could be found even within the dense core.16 Our primary goal in the present study is to test for the possible presence of elevated lipid in the vicinity of recently formed plaque in TgCRND8 and 3×Tg mice, and compare these with plaque from LOAD human tissue.The FTIR spectrum of normal grey matter is comprised of bands from all components: proteins, sugars, DNA, etc., as well as lipid membranes.12–16,23 The CH-stretch and the carbonyl stretch regions for normal grey matter and aggregated Aβ peptide16 are compared to spectra extracted from dense core plaques in each of the tissue samples in Fig. 1. The presence of lipid membrane can easily be illustrated by processing the data on the intensity of the CH2 symmetric stretch band, Fig. 1A. While there are likely some CH bonds in amino acid side groups, it can be seen that the band at 2850 cm−1 is essentially free of contributions from Aβ peptide. Similarly, the lipid carbonyl peak at 1738 cm−1 is absent in the fibrillar Aβ42. Spectra from the plaque cores from each type of sample invariably contain some CH2 symmetric stretch and lipid carbonyl, evidence of fatty acid ester lipids, such as comprise membranes. No core spectra could be found that did not contain these bands.
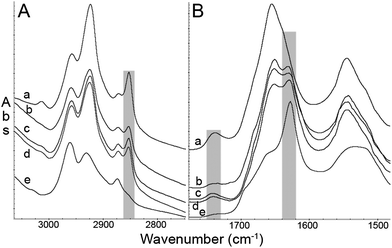 | ||
| Fig. 1 Representative FTIR spectra. (A) CH stretch region and (B) amide I and II region from (a) mouse grey matter, (b) human plaque core, (c) TgCRND8 mouse plaque core, (d) 3×Tg mouse plaque core, (e) fibrillar Aβ42. The symmetric CH2 peak, CO stretch peak, and β-sheet absorption in the amide I band are highlighted with grey boxes. With the exception of (e), spectra are displayed on a common scale, offset for clarity. | ||
Spectrochemical images of dense core plaques were acquired from 20 plaques in the 5 month and 16 in the 8 month TgCRND8 mice. The 3×Tg mice develop plaque at a later age. Three dense core plaques were imaged in the 12 month mice, 30 were imaged among the five 15 month animals and 8 in the two 17 month animals. Finally, 5 plaques were imaged from two brain sections from the human AD case. All spectrochemical images were processed for plaque and lipid. Representative processed images are shown in Fig. 2, 3 and 4 for TgCRND8, 3×Tg and human AD, respectively.
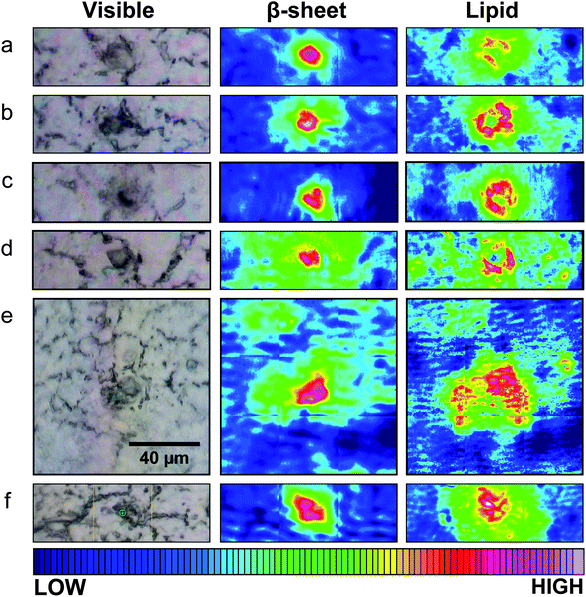 | ||
| Fig. 2 Typical visible and FTIR images of unstained tissue containing dense core plaque observed in three different CRND8 mice aged 5 months (a–d) and two aged 8 months (e and f). Left column: dense core appears darker than surrounding tissue under white light. Central column: aggregated plaque is shown from intensity of β-sheet shoulder. Right column: plaques are surrounded by lipid membrane-like signature that infiltrates to the core of all plaques, at this spatial resolution. Scale bar = 40 µm and applies to all images. Colour bar indicates low (blue) to high (red) for component imaged. | ||
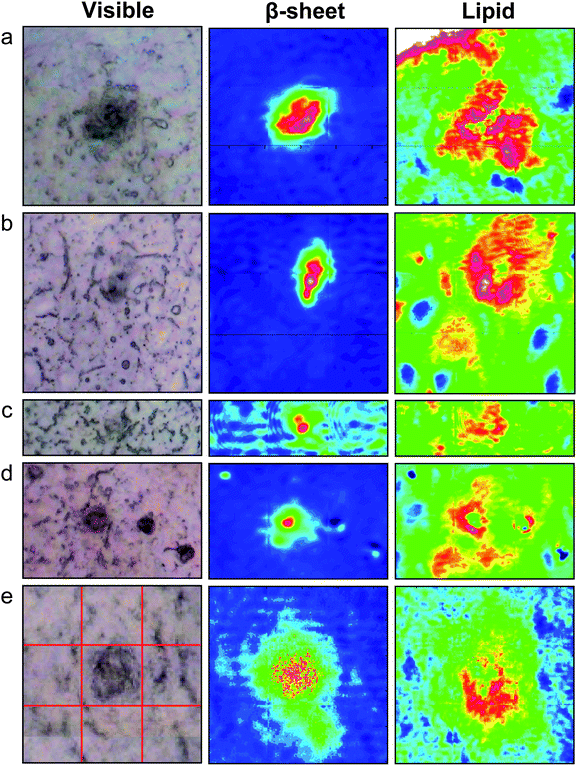 | ||
| Fig. 3 Typical visible and FTIR images of dense core plaque observed in five different 3×Tg mice aged (a) 12 months (b–d) 15 months and (e) 17 months. Left column: dense core appears darker than surrounding unstained tissue under white light. Central column: aggregated plaque is shown from intensity of β-sheet shoulder. Right column: plaques are surrounded by lipid membrane-like signature that infiltrates to the core of all plaques, at this spatial resolution. Red lines in (e) denote the 3 × 3 tiles of the mosaic. Tile = 64 × 64 pixels = 34.5 × 34.5 µm2. Colour scale as in Fig. 2. | ||
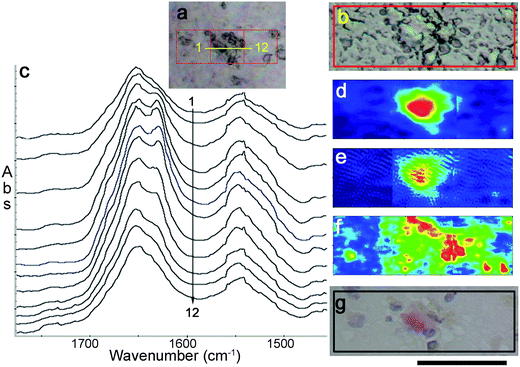 | ||
| Fig. 4 Images of dense core hippocampus plaque from a 91 year old AD patient. Visible images as viewed (a) through FTIR microscope (b) Nikon microscope. Spectra extracted from IRENI image along yellow line in (a) indicates location for (c) a stack of 12 spectra spanning the plaque. False-colour images of the 3 × 1 tile mosaic were processed to show (d) amide I band intensity due to aggregated β-sheet, (e) densest region of plaque core corresponding to a band at 1390 cm−1, and (f) symmetric CH2 stretch at 2850 cm−1 due primarily to lipids. Colour scale as in Fig. 2. (g) Photograph of tissue stained after FTIR imaging: plaque core (Congo red) and nuclei (Hematoxylin blue). Scale bar under (g) = 50 µm. | ||
For Fig. 2 and 3, a photoimage of each dense core plaque, as viewed under white light on the FTIR microscope, is shown next to the image processed for dense core plaque (intensity of β-sheet band) and for long chain fatty acid esters typical of lipid membrane. Dense core plaque scatters light, leading to a darkened area under white light illumination. The dense cores may be expected to act as scattering centers, and can lead to scattering artifacts. However, since the spectra are recorded in transmission, not transflectance, we have found that this distortion is minimal compared to the significant changes in the amide I and lipid bands. As has previously been shown,14–16 dense core plaque in TgCRND8 mice are invariably surrounded by a strong lipid signal, while even the core contains some lipid signature. The plaques in these younger mice have a similar structure, even at five months.
Remarkably, the dense core plaques from the 3×Tg mice exhibit these identical features, Fig. 3. In each case, the dense core plaque is located within a region that is high in lipid membrane relative to the surrounding region. Plaques in the 3×Tg mice first appear in the subiculum, and along the area of grey matter that lies between the alveus and the curve of the CA1 neurons.18 A plaque from a 12 month 3×Tg mouse (Fig. 3a), located in the latter region, is already in a lipid-rich environment. The upper left corner of the image includes the arc of the alveus, which is so rich in white matter tracts that the intensity is above the upper limits on our colour scale, and hence appears white. Nevertheless, the immediate vicinity of plaque shows relatively more intense lipid (red on the false colour scale).
The other 3×Tg plaques show similar lipid-rich environments: intense red surrounding plaque; regions beyond the plaque show as green, fading to blue in some cases. Cells in the subiculum region are scattered loosely, unlike the densely packed neurons of the CA; the cell bodies are also somewhat larger. With the increased spatial resolution provided by IRENI, the cell bodies in Fig. 3 (especially in a, b and d) appear as dark blue (low lipid) ovals, easily distinguished from surrounding neuropil and white matter tracts under this lipid processing.16
One of the five plaques imaged from the human AD case is shown in Fig. 4. Scatter from the dense tissue creates some dark shapes in the FTIR white light image, Fig. 4a, but not all are from plaque core. A 40× image from a Nikon Alphaphot-2 microscope is shown in Fig. 4b, for better appreciation of the unstained tissue morphology. Twelve spectra extracted from along a line across the plaque (total length = ∼75 pixels) illustrate the extent of the fibrillar Aβ distribution in this plaque, Fig. 4c. Since spatial resolution is on the order of 1–2 µm for CH stretch wavelength range, while the plaque extends across several 10's of µm, it is evident that some lipid membrane material lies within the plaque core region. The spectrochemical image processed for the β-sheet band at 1630 cm−1 (Fig. 4d) shows that some Aβ signature can be detected all across the center tile. The densest Aβ aggregation can be illustrated by the intensity of a band centered at 1390 cm−1,16Fig. 4e. This band is co-located with the lowest lipid distribution, Fig. 4f, but some lipid is present even here. The elevated lipid signature is seen throughout the entire plaque, highest at the periphery but infiltrating the total plaque area in a manner that closely mimics the results found in the transgenic mice. Location of the dense core is confirmed in the Congo red stain (Fig. 4g). The periphery of the plaque stains faintly with Congo red, where the spectral signatures show that it incorporates a greater amount of lipid than the core (Fig. 4d–g). All plaques from this sample had a similar aspect.
Discussion
The major finding in this paper is that our spectrochemical image analysis shows a lipid membrane-like signature surrounding and infiltrating the dense core plaques not only in TgCRND8 mouse brain, as had been reported by us earlier,14–16 but also in early stage plaque in the 3×Tg mouse brain and in plaque from post mortem ApoE43 human brain at Braak stage 6 AD. We now discuss this feature and possible explanations for its presence.In Fig. 1, the well-known CH2 and CH3 motif is accompanied by lipid carbonyl, leading to the assignment as long chain fatty acid esters, which comprise lipid bilayer membranes. A small band, at about 3012 cm−1, can also be seen in the spectrum of normal grey matter (Fig. 1) and traces of it are evident in the three plaque spectra. Comparisons among spectra from dried and freshly thawed tissues show that certain restrictions in tissue preparation and storage are critical for preservation of some chemically unstable biomarkers, in particular unsaturated CC and to some extent the lipid carbonyl CO.19 Our protocols were designed to ensure that all samples were maintained frozen until just prior to analysis. From the data, we are confident that the spectral signatures represent the state of the tissue at acquisition, without any degradation in lipid signature. Moreover, we can deduce that the lipid composition around the plaque contains some small amount of unsaturated fatty acid esters, but nothing out of proportion to normal grey matter composition.
The spectrochemical analysis indicates that lipid is a common feature of the physical structure of the plaques, regardless of the driving forces or genetic causes for disease development. Hypotheses concerning the trigger events and driving forces in the development of AD remain unresolved. This lack of clarity arises from the complexity of the disease, which is widely regarded to be multifactorial in origin, and has a tremendously complex etiology. Among several plausible explanations for the appearance of this lipid signature, we note the following:
(1) lipid membrane surfaces may play a role in templating the aggregation of oligomers into β-sheets, inducing fibrillogenesis.24,25
(2) Lipid may play a different pathogenic role, reversing the dense core plaque into more toxic, soluble protofibrils.26
(3) Microglia are well-known to surround and infiltrate plaque, in their phagocytotic role of sequestering Aβ; hence the lipid signature could arise simply from the presence of a quantity of long processes associated with activated microglia.27,28
(4) Astrocytes have recently been shown to attenuate plaque formation APP/PS1 mouse.29 Like microglia, these cells are characterized by small cell bodies and numerous star-like processes that could offer a lipid-rich signature in the IR spectrum.
(5) Apolipoprotein E can also bind Aβ for clearance from the brain. This process, particularly though the pro-neurotrophin receptor sortilin, may be disrupted in AD.30
Of the five possibilities mentioned here, it is difficult to ascertain which may be more likely. Both (1) and (2) propose lipid roles in membrane aggregation/dissolution. In this regard, we note that 3D electron microscopy reconstruction suggests a close association between Aβ and dendrosomatic membranes in 3×Tg mice and aged dogs.31 The regions that appear blue-green (low lipid) in Fig. 2–4 represent normal grey matter: larger cell bodies appear as blue ovals, neuropil is blue to green. Explanations (3) and (4) both ascribe the elevated lipid signature to processes from cells; this would require that there be an unusually dense accumulation of such processes. Since both types of cell will naturally migrate to sites of injury, and do accumulate around plaque, this remains a viable possibility. Dystrophic neurites might also contribute. Finally, apolipoprotein E is a major transporter of lipids; its presence could account in part for the elevated lipid signature.
It is not our intention to try to sort through these and other competing/contributing factors in the evolution of AD. Suffice to note that there is ample evidence to support the validity of our observations as being closely related to plaque evolution, and possibly to common steps in aggregation and/or clearance of Aβ.
The visible image in Fig. 3d shows four dark spots but only the central one is an amyloid plaque; the remainder are small crystals of creatine, formed during the sectioning and drying process. In our earlier studies on mouse brain,13,15 we employed a different method for snap-freeze (immersion of OCT-coated sample directly into liquid N2) which may have contributed to the streak-like appearance of most creatine crystals. In spectrochemical imaging, we need both high & low spatial resolution: it is necessary to match pixel size to the question. In the case of creatine deposits, because they are highly localized, it is critical to survey the entire region of interest (hippocampus, cortex, caudate, etc.) in a statistically significant number of cases. This study is best suited to large surveys performed with a standard thermal source FTIR microscope with FPA. We are investigating the possibility of neuroprotective strategies that may involve creatine or creatine kinase, but this investigation requires a comprehensive overview of tissues and a full suite of age-matched controls. In contrast, the observations reported here, and the information gleaned from this first analysis, are made possible only because of the spatial resolution and remarkably clear spectral data that can be obtained with the IRENI system. To assist in future analysis, we are developing sub-space clustering and frequent pattern mining methods. In addition, we have not yet applied spatial deconvolution methods32 to the IRENI data. The challenge now lies in obtaining the maximum amount of information possible, using only the biochemical signatures of naturally occurring components in unstained, unfixed tissue to explore the myriad unknowns within these rich spectrochemical data sets.
Conclusions
Synchrotron FTIR imaging of brain sections, from two different transgenic mouse models of AD (TgCRND8 and 3×Tg) and from human LOAD, shows that all dense core plaques are surrounded and infiltrated with lipid membrane components. The spatial resolution afforded with the IRENI system shows intimate integration of membrane and aggregated Aβ at 1 µm length scale, throughout plaques that are >20 µm in diameter. Several possible explanations for this are proposed, including lipid as a template for aggregation and/or dissolution of plaque, increased numbers of inflammatory response cells (microglia and astrocytes) and apolipoprotein E; further tests are on-going.Acknowledgements
The authors are grateful to Dr David Westaway for the original donation of TgCRND8 mice, and to Alex Paterson and Scott McKay for technical assistance. This work was supported by grants from NSERC Canada (BCA, KMG). Dr Benedict Albensi holds The Honourable Douglas Everett, Patricia Everett and the Royal Canadian Properties Endowment Fund Chair and is a Research Affiliate at the Centre on Aging, University of Manitoba. The IRENI project (CJH) is supported by NSF Grant no. DMR-0619759. The SRC is operated under NSF Grant no. DMR-0537588.Notes and references
- D. J. Selkoe, Physiol. Rev., 2001, 81, 741–766 CAS.
- Alzheimer's Society of Canada, Rising tide: the impact of dementia on Canadian society, 2010, isbn 978-0-9733522-2-1 Search PubMed.
- C. R. Jack, M. S. Albert, D. S. Knopman, G. M. McKhann, R. A. Sperling, M. C. Carrillo, B. Thies and C. H. Phelps, Alzheimer's Dementia, 2011, 7, 257–262 CrossRef.
- G. M. McKhann, D. S. Knopman, H. Chertkow, B. T. Hyman, C. R. Jack, C. H. Kawas, W. E. Klunk, W. J. Koroshetz, J. J. Manly and R. Mayeux, et al. , Alzheimer's Dementia, 2011, 7, 263–269 CrossRef.
- M. S. Albert, S. T. DeKosky, D. Dickson, B. Dubois, H. H. Feldman, N. C. Fox, A. Gamst, D. M. Holtzman, W. J. Jagust and R. C. Petersen, et al. , Alzheimer's Dementia, 2011, 7, 270–279 CrossRef.
- R. A. Sperling, P. S. Aisen, L. A. Beckett, D. A. Bennett, S. Craft, A. M. Fagan, T. Iwatsubo, C. R. Jack, J. Kaye and T. J. Montine, et al. , Alzheimer's Dementia, 2011, 7, 280–292 CrossRef.
- T. Gomez-Isla, R. Hollister, H. West, S. Mui, J. H. Growdon, R. C. Petersen, J. E. Parisi and B. T. Hyman, Ann. Neurol., 1997, 41, 17–24 CrossRef CAS.
- D. A. Bennett, J. A. Schneider, R. S. Wilson, J. L. Bienias and S. E. Arnold, Arch. Neurol., 2004, 61, 378–384 CrossRef.
- P. T. Nelson, E. L. Abner, F. A. Schmitt, R. J. Kryscio, G. A. Jicha, C. D. Smith, D. G. Davis, J. W. Poduska, E. Patel, M. S. Mendiondo and W. R. Markesbery, Brain Pathol., 2010, 20, 66–79 CrossRef.
- E. Karran, J. Neurochem., 2012, 123, 647–651 CrossRef CAS.
- R. W. Mahley and Y. Huang, Neuron, 2012, 76, 871–885 CrossRef CAS.
- K. M. Gough, M. Rak, A. Bookatz, M. Del Bigio, S. Mai and D. Westaway, Vib. Spectrosc., 2005, 38, 133–141 CrossRef CAS.
- M. Gallant, M. Rak, A. Szeghalmi, M. R. Del Bigio, D. Westaway, J. Yang, R. Julian and K. M. Gough, J. Biol. Chem., 2006, 281, 5–8 CrossRef CAS.
- M. Rak, M. R. Del Bigio, S. Mai, D. Westaway and K. M. Gough, Biopolymers, 2007, 87, 207–217 CrossRef CAS.
- A. Kuzyk, M. Kastyak, V. Agrawal, M. Gallant, G. Sivakumar, M. Rak, M. R. Del Bigio, D. Westaway, R. Julian and K. M. Gough, J. Biol. Chem., 2010, 285, 31202–31207 CrossRef CAS.
- M. Z. Kastyak-Ibrahim, M. J. Nasse, M. Rak, C. Hirschmugl, M. R. Del Bigio, B. C. Albensi and K. M. Gough, NeuroImage, 2012, 60, 376–383 CrossRef CAS.
- M. A Chishti, D. Yang, C. Janus, A. L. Phinney, P. Horne, J. Pearson, R. Strome, N. Zuker, J. Loukides, J. French, S. Turner, G. Lozza, M. Grilli, S. Kunicki, C. Morissette, L. Paquette, F. Gervais, C. Bergeron, P. E. Fraser, G. A. Carlson, P. St. George-Hyslop and D. Westaway, J. Biol. Chem., 2001, 276, 21562–21570 CrossRef.
- S. Oddo, A. Caccamo, M. Kitazawa, B. P. Tseng and F. M. LaFerla, Neurobiol. Aging, 2003, 24, 1063–1070 CrossRef CAS.
- D. M. Stitt, M. Z. Kastyak-Ibrahim, C. R. Liao, J. Morrison, B. C. Albensi and K. M. Gough, Vib. Spectrosc., 2012, 60, 16–22 CrossRef CAS.
- H. Braak and E. Braak, Acta Neuropathol., 1991, 82, 239–259 CrossRef CAS.
- M. J. Nasse, R. Reininger, T. Kubala, S. Janowski and C. J. Hirschmugl, Nat. Methods, 2011, 8, 413–416 CrossRef CAS.
- C. J. Hirschmugl and K. M. Gough, Appl. Spectrosc., 2012, 66, 475–491 CrossRef CAS.
- J. M. Chalmers and P. R. Griffiths, Handbook of Vibrational Spectroscopy, Wiley, Chichester UK, 2002, vol. 1–5 Search PubMed.
- M. Bokvist, F. Lindstrom, A. Watts and G. Grobner, J. Mol. Biol., 2004, 335, 1039–1049 CrossRef CAS.
- E. Y. Chi, C. Ege, A. Winans, J. Majewski, G. Wu, K. Kjaer and K. Y. C. Lee, Proteins, 2008, 72, 1–24 CrossRef CAS.
- I. C. Martins, I. Kuperstein, H. Wilkinson, E. Maes, M. Vanbrabant, W. Jonckheere, P. Van Gelder, D. Hartmann, R. D'Hooge, B. De Strooper, J. Schymkowitz and F. Rousseau, EMBO J., 2008, 27, 224–233 CrossRef CAS.
- M. Kitazawa, S. Oddo, T. R. Yamasaki, K. N. Green and F. M. LaFerla, J. Neurosci., 2005, 25, 8843–8853 CrossRef CAS.
- I. Luccarini, C. Grossi, C. Traini, A. Fiorentini, T. E. Dami and F. Casamenti, Neurosci. Lett., 2012, 506, 94–99 CrossRef CAS.
- A. W. Kraft, X. Hu, H. Yoon, P. Yan, Q. Xiao, Y. Wang, S. C. Gil, J. Brown, U. Wilhelmsson, J. L. Restivo, J. R. Cirrito, D. M. Holtzman, J. Kim, M. Pekny and J. Lee, FASEB J., 2013, 27, 187–198 CrossRef CAS.
- A. Carlo, C. Gustafsen, G. Mastrobuoni, M. S. Nielsen, T. Burgert, D. Hart, M. Rohe, A. Nykjaer, J. Herz, J. Heeren, S. Kempa, C. M. Petersen and T. E. Willnow, J. Neurosci., 2013, 33, 358–370 CrossRef CAS.
- P. Nuntagij, S. Oddo, F. M. LaFerla, N. Kotchabhakdi, O. P. Ottersen and R. Torp, J. Alzheimer's Dis., 2009, 16, 315–323 CAS.
- E. C. Mattson, M. J. Nasse, M. Rak, K. M. Gough and C. J. Hirschmugl, Anal. Chem., 2012, 84, 6173–6180 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |