Disposable screen-printed sensors for the electrochemical detection of TNT and DNT†
J. Sarah
Caygill
a,
Stuart D.
Collyer
b,
Joanne L.
Holmes
a,
Frank
Davis
a and
Séamus P. J.
Higson
*a
aCranfield Health, Vincent Building, Cranfield University, Bedfordshire, MK43 0AL, UK. E-mail: s.p.j.higson@cranfield.ac.uk; Fax: +44 (0)1234-758380; Tel: +44 (0)1234-758516
bMicroarray Ltd, PO Box 88, Manchester, M60 1QD, UK
First published on 7th November 2012
Abstract
Due to the heightened level of national security currently prevalent due to the possibility of terrorist incidents, highly portable, miniaturised and sensitive monitoring devices for trace levels of injurious materials, such as explosives are now of the upmost importance. One method that offers a possible route for the development of a detection system for such species is via an electrochemical regime, coupled to the use of disposable sensor technology. Within this study, the use of carbon screen-printed sensors for the detection and analysis of the classical explosive trinitrotoluene (TNT) and the related dinitrotoluene (DNT) is described, with the eventual objective to develop an inexpensive, accurate and sensitive detection system for trace quantities of explosives in field settings. Commercially available screen-printed carbon sensors have been used as the base platform for this investigation and the electrochemistry of both TNT and DNT studied at these surfaces. Two reductive peaks and one oxidative peak were observed for both analytes. The best linear fits and sensitivities were obtained using the reductive peak at −0.72 V vs. Ag/AgCl. A linear range from 1 to 200 μM could be obtained for TNT and DNT in pH 7.0 phosphate buffer with limits of detection as low as 0.4 μM (TNT) and 0.7 μM (DNT). A second system which utilised the addition of the enzyme, nitroreductase, and the coenzyme, NADPH, into the solution matrix prior to electrochemical interrogations with screen-printed carbon electrodes was found to increase the resulting signal magnitude at the oxidation peak at +0.3 V, improving the performance of the sensor at these values.
Introduction
The increased concern for homeland security has led to an upsurge in research into a wide range of systems capable of detecting materials capable of causing widespread public harm such as explosives, chemical or biological weapons at particularly low levels. A number of commercial systems presently exist, usually based on some form of mass spectroscopy, which can detect such compounds at these low levels. However these systems are both expensive and often bulky. There has been much interest in the development of more compact and less costly methods for the detection of compounds, such as TNT, for ‘in-field’ application.1,2Electrochemical sensors have been widely used to detect many explosive-related analytes of interest and display high sensitivities and selectivities, especially when combined with the use of biological molecules with highly specific recognition or catalytic properties to make biosensors. Electrochemical sensors however are usually suitable for detection of analytes only when in solution. This does not mean however, that they cannot be applied for the detection of explosive material, and for example buried ordinance could well contaminate groundwater which could be analysed by these sensors. Alongside this, swabbing of suspects hands combined with ELISA assays is often used to investigate whether they have recently handled explosives, however ELISA assays can be time consuming, costly and require trained personnel; a simple sensor-based device could reduce the time and cost of this procedure. Also there is the potential for combining the electrochemical sensor with an air extraction system which could concentrate airborne analytes into solution and allow their detection. For example early work combined a commercial cyclone air extractor which extracted TNT vapour from the air and concentrated it into distilled water. The resulting aqueous sample was then passed over an immobilised monoclonal antibody to TNT, displacing a fluorescent marker compound from the antibody.3 This system proved capable of detecting trace amounts of TNT in air.
There have been a number of electrochemical sensors developed for a range of explosives, including TNT.1,2 TNT can be detected electrochemically since it undergoes a number of electrochemical transformations,4 which can be related back to the concentration being evaluated. A number of carbon and gold screen-printed electrodes have been used to detect the voltammetric reduction of nitro groups in TNT with detection limits of 200 μg L−1 having been reported.5 By combining the use of carbon micro-fibre electrodes with square-wave voltammetry, electrochemical sensors for nitroaromatic compounds, including TNT, were developed with detection limits of approximately 30 μg L−1.6 Other workers coated a screen-printed carbon electrode with an agarose hydrogel; exposure of this sample to TNT vapour led to its adsorption into, and therefore concentration by, the hydrogel.7 Square-wave voltammetry was then used to detect the vapour given off by a solid sample of TNT. Nafion has also been used to coat screen-printed carbon electrodes, with these being then used to analyse solutions of nitroaromatic compounds and successfully detected 30 μmol L−1 TNT in spiked lake water.8 Various other materials have also been incorporated into sensors, such as mesoporous silica which was immobilised onto glassy carbon by a layer-by-layer approach to give a sensor capable of detecting 1.5 nmol L−1 of TNT in water.9 Combining silica with ordered mesoporous carbon also gave a sensor with a low detection limit; 200 ng L−1.10
Other techniques have included incorporating mercury/gold amalgam electrodes into a flow through system11 to give high throughput measurements with a detection limit of 7 μg L−1 or depositing carbon screen-printed electrodes onto a Gore-Tex fabric to give wearable electrochemical sensors for TNT.12 Furthermore, recently nanostructured materials have been utilised within these sensors, carbon nanotube (CNT)–metal nanoparticle (NP) composites have been investigated with a CNT–Cu NP composite allowing detection of TNT by adsorptive stripping voltammetry13 at levels as low as 1 mg L−1, whereas other workers combined CNTs with titanium dioxide and metal nanoparticles14 and showed enhanced activity. Graphene has also recently been of interest, with graphene and graphite nanoparticles both being used in electrochemical sensors for TNT, with detection limits of 1 μ L−1 in seawater,15 whereas other workers could detect 42 nmol L−1 levels of DNT using electrochemically reduced graphene16 and ionic liquid–graphene composites have also been utilised with detection limits for TNT as low as 4 μg L−1 in sea, lake or tap water.17
Within this work we have attempted to develop and characterise a much simpler system based on inexpensive screen-printed electrodes without the need for complex nanomaterials or assembly procedures for the detection of aqueous nitro-explosives. Our system will allow for the development of inexpensive single-shot sensors for TNT or similar compounds. Also since other workers have reported the use of enzymes in biosensors for TNT18,19 with high sensitivity and low detection limits (2 μmol L−1) we also investigated the effect of adding a nitroreductase enzyme and its co-factor to our system in an attempt to improve sensitivity and linear range.
Materials and methods
Reagents and materials
Sodium dihydrogen phosphate 1-hydrate, disodium hydrogen phosphate 12-hydrate and sodium chloride (all AnalaR grade) were obtained from BDH Laboratory Supplies (Poole, Dorset, UK).2,4,6-Trinitrotoluene (1000 μg mL−1 in acetonitrile) and 2,4-dinitrotoluene (1000 μg mL−1 in acetonitrile) were obtained from Supelco (Bellefonte, Pennsylvania, USA). Acetonitrile (AnalaR), β-nicotinamide adenine dinucleotide 2′-phosphate NADPH (reduced tetrasodium salt) and nitroreductase (type nfsA from Escherichia coli) were obtained from Sigma (Poole, Dorset, UK). Sulphuric acid (98%) and hydrogen peroxide (30%) were purchased from Fisher Scientific (Loughborough, Leicester, UK).
All solutions were prepared with deionised water (≥18 MΩ cm−1 resistivity) from an Elga Purelab UHQ-II Water System (Vivendi Water Systems, High Wycombe, Buckinghamshire, UK). A pH 7.0 phosphate buffer solution was prepared, comprising: 4.3 × 10−3 M sodium dihydrogen phosphate, 5.6 × 10−3 M disodium hydrogen phosphate and 7.87 × 10−2 M sodium chloride.
A Piranha solution consisting of a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 combination of 98% sulphuric acid and 30% hydrogen peroxide was used for the removal of organic residues from the glass cell used in electrochemical interrogations between experimental procedures. (Caution – Piranha is highly oxidising and corrosive.)
:1 combination of 98% sulphuric acid and 30% hydrogen peroxide was used for the removal of organic residues from the glass cell used in electrochemical interrogations between experimental procedures. (Caution – Piranha is highly oxidising and corrosive.)
The screen printed carbon electrodes (SPCE) were obtained from Microarray (Manchester, UK) and comprised of a 4-electrode system with two carbon ink working electrodes (electroactive area of 22 mm2 per WE), a carbon ink counter electrode and a silver/silver chloride reference electrode, as depicted in Fig. S1 (ESI†). These were purchased in sheets of 45 (Fig. S1†) and were used individually as required (Fig. S2†).
Experimental apparatus
An analytical electrochemical workstation (PG580) from Uniscan Instruments (Buxton, UK) was used for all electrochemical studies, the current/charge outputs resulting from sensor interrogation were recorded with the relevant software supplied with the workstation.Sealed electrochemical cell
A sealed prototype electrochemical cell was designed in-house and fabricated by Soham Scientific (Ely, Cambridge, UK) for use in all electrochemical studies (Fig. S3 and S4, ESI†).Electrochemical interrogation with a screen printed carbon sensor
Cyclic voltammetry interrogations were performed on a single WE of the screen-printed sensors in the presence of TNT/DNT (and when required in the presence of TNT with the enzyme nitroreductase and the cofactor NADPH) in a sealed glass cell. The solutions of DNT/TNT, with/without nitroreductase and NADPH were all prepared in the pH 7.0 phosphate buffer. Prior to electrochemical interrogation, the cell was deoxygenated by purging with argon (BOC, Letchworth, UK) gently for ten minutes. The cyclic voltammetric investigation of nitro compounds was performed by sweeping the potential between −0.8 and +0.8 V (vs. Ag/AgCl) for one cycle. Unless otherwise stated, the scan rate for all interrogations was 25 mV s−1.Results and discussion
Cyclic voltammetry of TNT and DNT
When a potential sweep is applied to the electrode between −0.8 and +0.8 V (vs. Ag/AgCl) within a solution containing 200 μM TNT or DNT (Fig. 1), significant peaks in the current response are observed. Anodic and cathodic peaks are apparent indicating both oxidative and reductive reactions are taking place. Nitroaromatics have been observed to be electroactive compounds1,4,5 to the inherent redox activity caused by the easily reducible nitro groups behaving as electron acceptors. | ||
| Fig. 1 Cyclic voltammogram of 200 μM TNT or DNT in a solution of pH 7.0 phosphate buffer solution (vs. Ag/AgCl) at a scan rate of 20 mV s−1. | ||
Taking the electrochemical profile of TNT first (Fig. 1), the reductive peak (R1) present at a potential of −0.72 V (vs. Ag/AgCl) is indicative of the electrochemical reduction of the nitro groups on the TNT molecule as a result of a sequential four-electron reduction of each nitro group to its hydroxylamine derivative (Fig. 2a).8
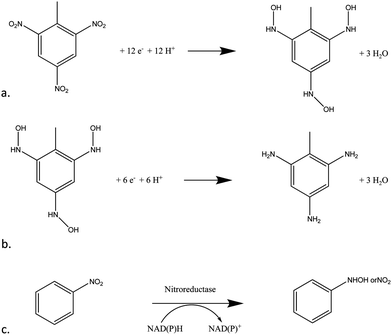 | ||
| Fig. 2 (a) Four-electron reduction of nitro groups on TNT (b) two-electron reduction of TNT's nitro groups from hydroxylamine to amine (c) the enzymatic reaction of nitroreductase and the cofactor NADPH on a molecule with a nitro moiety. | ||
The mechanism by which this reduction takes place on poly-nitroaromatic compounds depends on several factors including the number of nitro groups, their relative position on the ring, the nature of other substituents on the ring and the solution pH.4 The electron deficient nitrogen on the nitro groups acts as an electron acceptor thus enabling the four-electron reduction to take place at each nitro group (Fig. 2a). Once the reduction from nitro to hydroxylamine has taken place, a reversible second step, from hydroxylamine to amine occurs with a two-electron reduction (Fig. 2b). This can be observed as an anodic and cathodic peak redox couple at +0.25 V (O1) and −0.30 V (R2).
For DNT, the mechanism of reduction is largely the same – with the two nitro groups being reduced first to hydroxylamine and then reversibly to amine moieties. It is interesting to note that DNT is less easily reduced than TNT and so the position of the reduction peaks should occur at a more negative potential.1 It can be seen that the peak positions of DNT and TNT are similar but not in precisely the same position – there is an approximate 50 mV (vs. Ag/AgCl) shift in the cathodic direction in the DNT voltammograms when compared to those obtained of TNT.
Effect of oxygen
It is suggested that in this instance the reduction of the nitro-group at −0.72 V will be the more practical to monitor, since the presence of reducible nitro groups is rare in nature. Other workers have also preferred this transition for quantitative detection.1 However one potential interferent at these lower potentials is oxygen, and therefore we attempted to quantify this effect by running voltammograms of 100 μmol L−1 TNT solutions which had been de-oxygenated with argon for time periods of 0–20 minutes. Fig. 3 exhibits some of the voltammograms obtained using this process, and as can be seen initially there is the large peak again at −0.72 V which decreases in magnitude following purging for 2 and 4 minutes. The inset shows the peak current value obtained at −0.72 V against de-oxygenation time, and as can be seen after 4 minutes there is little further change in this value, therefore a de-oxygenation time of 10 minutes was adopted as a standard step prior to all further electrochemical investigations. The peaks at +0.25 and −0.30 V were much less affected by the presence of oxygen (Fig. 3).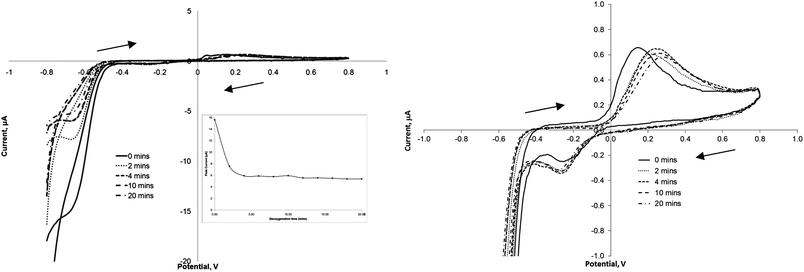 | ||
| Fig. 3 Cyclic voltammograms representing the increase in argon purging time and the influence this has on the oxidative and reductive peaks of 100 μM TNT (in phosphate buffer at a scan rate of 20 mV s−1. Inset: plot of peak current at −0.72 V vs. Ag/AgCl vs. purging time. | ||
Calibration curves
A range of differing concentrations of TNT were then examined by cyclic voltammetry following 10 min de-oxygenation and the currents obtained at all three redox peaks determined. Fig. 4 shows the voltammograms obtained for concentrations of 0, 50, 100 and 200 μmol L−1 TNT. The resulting peak current for each distinct redox peak against exposed concentration are shown in Fig. 5. For the peak currents obtained at −0.72 V there appears to be an extremely good linear response (R2 = 0.9986), the peak currents at −0.30 V also exhibits a similar linearity (R2 = 0.9996) although the response is less sensitive due to a smaller peak height. Interestingly the calibration plot for the oxidative peak at +0.25 V shows the on-set of a plateau at 200 μmol L−1. | ||
| Fig. 4 Cyclic voltammograms of increasing concentrations of TNT in phosphate buffer solution (scan rate 20 mV s−1vs. Ag/AgCl). | ||
 | ||
| Fig. 5 Calibration plots for TNT in phosphate buffer solution on a screen-printed carbon electrode taken from the peak in position at +0.25 V, −0.30 V and −0.72 V (vs. Ag/AgCl) – standard deviations (n = 3) are shown. | ||
Lower levels of TNT were also utilised to attempt to determine the limits of detection of this system. Again the peak at −0.72 V displays a good linear range from 0–40 μmol L−1 TNT (R2 = 0.9980) with a limit of detection (determined from 3× the standard deviation of the baseline value) of 0.4 μmol L−1, corresponding to approximately 90 μg L−1. Similar results could also be obtained with the other two peaks, however both of these led to lower degrees of linearity, relatively larger standard deviations and higher limits of detection. These results are summarised in Table 1.
| Detection limit in μmol L−1 | |||
|---|---|---|---|
| R1 | R2 | O1 | |
| TNT | 0.4 | 2.6 | 5.0 |
| DNT | 0.7 | 7.5 | 7.0 |
| TNT/NTR + NADPH | 0.5 | 20.0 | 15.0 |
An identical sequence of experiments were carried out using DNT. All three peak currents again led to high degrees of linearity between 0 and 200 μmol L−1 with R2 > 0.997 (Fig. 6). For the major reduction peak (−0.72 V) the sensitivity (determined from the slope of the calibration profile) appeared to be slightly higher than for TNT at greater concentrations levels, however at lower levels the response was observed to diminish. Lowered sensitivity was also again noted for the other two peaks (O1) and (R2). It is suggested here that this is related to a combination of the number of nitro groups available (2 vs. 3) per molecule and also the relative rate of each electrochemical transformation. Others20 have also shown an increased DNT peak current response in comparison to TNT when tested comparatively at 50 μmol L−1. Sensitivity in this instance may be enhanced by decreasing the scan rates currently employed – although this in turn could increase system response times. Limits of detection appear to be higher for DNT, as a result of decreased sensitivity at lowered concentration levels (Table 1).
 | ||
| Fig. 6 Calibration plots for DNT in phosphate buffer solution on a screen-printed carbon electrode taken from the peak in position at +0.20 V, −0.30 V and −0.77 V (vs. Ag/AgCl) – standard deviations (n = 3) are shown. | ||
Effect of nitroreductase
Fig. 2c shows the schematic catalytic reduction of nitro compounds by nitroreductase (NTR) enzymes and corresponding oxidation of the co-factor NADPH. Both these materials were added to the TNT solutions to ascertain if they could act as mediators and therefore improve the rate of reaction and correspondingly lower detection limits. Cyclic voltammograms of 200 μmol L−1 TNT were compared (Fig. S5, ESI†) and it may be observed that the voltammogram obtained from a solution containing the enzyme and cofactor is almost identical in shape to that attained without the enzyme and cofactor present. Fig. 7 shows a calibration plot for TNT solutions containing 20 units nitroreductase and 0.1 mM NADPH, again as can be seen there appears to be little advantage in utilising this system except that the oxidative peak at +0.30 V has now shifted to +0.25 and also exhibited an increased peak current response by up to 50% at this higher concentration of TNT employed. It is also worth noting that peak current values for R1 and O1 are also lowered. It is suggested here that, using the concentrations employed here for NTR and NADPH, there is in fact a decrease in the amount of reduced –NO2 groups in the system (as evidenced by the decreased peak area of the −0.72 V peak for the enzymatic system), and therein decreasing the response of the oxidation/reduction linked reaction. Standard deviations also appear to be larger in this more complex system, leading to increased limits of detection (Table 1). It is worth noting here however that these are preliminary results and investigations into the effects of varying the concentrations of NTR/NADPH on signal response are on-going and will be included in future publications.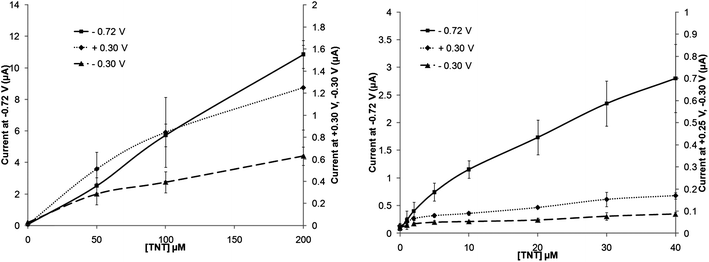 | ||
| Fig. 7 Calibration plot of increasing TNT concentrations with 20 units nitroreductase and 0.1 mM NADPH (taken at a scan rate of 20 mV s−1) – standard deviations (n = 3) are shown. | ||
Selectivity and applicability of sensors to “real” environmental samples is an important factor in their deployment. Although a simple potentiostatic experiment could struggle to distinguish DNT and TNT from interferents undergoing reduction reactions at the same potentials, the use of cyclic voltammetry does however allows differentiation between DNT and TNT as shown in Fig. 1, with the peaks R1 and O1 occurring at differing potentials for both species. Work reported by other authors6,8 also demonstrates differentiation between explosive compounds such as DNT/TNT from others such as nitrobenzene. Electrochemical detection of nitro-explosives has also been previously demonstrated in environmental samples8,15,17 and in on-going work by ourselves using mediated electrode systems. These displayed excellent differentiation between differing nitroaromatic species, as well as demonstrating detection of environmental and in particular atmospheric-born samples of materials such as DNT and TNT which will be reported in-further publications.
Conclusions
We have demonstrated the use of inexpensive, commercial screen-printed electrodes as a suitable platform for the electrochemical detection of both TNT and DNT. The most reliable and sensitive method appeared to be monitoring the reduction reaction that occurs at −0.72 V vs. Ag/AgCl. Limits of detection as low as 0.4 μmol L−1 could be achieved with linear ranges from at least 1–200 μmol L−1 obtained (R2 = 0.998). Addition of a nitroreductase enzyme and NADPH cofactor to the system had minimal effect on the reductive electrochemical transitions and at the concentrations employed in this study, caused a measurable decrease in current response to the oxidative signal responses observed.Whilst the sensing method described herein is not as sensitive as others previously reported in the literature at present, the use of commercial screen-printed electrodes coupled to a simplistic electrochemical detection method would allow large numbers of tests to be run under field conditions, perhaps in determining the levels of environmental pollution by stored or buried ordnance or detecting explosive residues in swabbed samples. Alternatively it may be possible to combine this method with air sampling techniques which could be used to concentrate explosive vapours and allow their detection in atmospheric samples. It is also worth noting that results reported here are preliminary and working is on-going involving both chemical and biochemical modifications of the base electrode substrate to increase both sensor selectivity and specificity.
Acknowledgements
The authors would like to acknowledge the Engineering and Physical Sciences Research Council (EPSRC) for the funding and sponsorship of an Eng.D studentship for J.S.C.References
- J. Wang, Electroanalysis, 2007, 19, 415–423 CrossRef CAS.
- J. S. Caygill, F. Davis and S. P. J. Higson, Talanta, 2012, 88, 14–29 CrossRef CAS.
- J. P. Whelan, A. W. Kusterbeck, G. A. Wemhoff, R. Bredehorst and F. S. Ligler, Anal. Chem., 1993, 65, 3561–3565 CrossRef CAS.
- K. Bratin, P. T. Kissinger, R. C. Briner and C. S. Bruntlett, Anal. Chim. Acta, 1981, 130, 295–311 CrossRef CAS.
- J. Wang, F. Lu, D. MacDonald, J. Lu, M. E. S. Ozsoz and K. R. Rogers, Talanta, 1998, 6, 1405–1412 CrossRef.
- L. Aguí, D. Vega-Montenegro, P. Yáñez-Sedeño and J. M. Pingarrón, Anal. Bioanal. Chem., 2005, 382, 381–387 CrossRef.
- K. Cizek, C. Prior, C. Thammakhet, M. Galik, K. Linker, R. Tsui, A. Cagan, J. Wake, J. L. Belle and J. Wang, Anal. Chim. Acta, 2010, 661, 117–121 CrossRef CAS.
- J. Chen, J. Shih, J. C. Liu, M. Kuo and J. Zen, Anal. Chem., 2006, 78, 3752–3757 CrossRef CAS.
- G. Shi, Y. Qu, Y. Zhai, Y. Liu, Z. Sun, J. Yang and L. Jin, Electrochem. Commun., 2007, 9, 1719–1724 CrossRef CAS.
- J. Zang, C. X. Guo, F. Hu, L. Yu and C. M. Li, Anal. Chim. Acta, 2011, 683, 187–191 CrossRef CAS.
- J. Wang and M. Pumera, Talanta, 2006, 69, 984–987 CrossRef CAS.
- M. Chuang, J. R. Windmiller, P. Santhosh, G. V. Ramírez, M. Galik, T. Chou and J. Wang, Electroanalysis, 2010, 22, 2511–2518 CrossRef CAS.
- S. Hrapovic, E. Majid, Y. Liu, K. Male and J. H. T. Luong, Anal. Chem., 2006, 78, 5504–5512 CrossRef CAS.
- B. Filanovsky, B. Markovsky, T. Bourenko, N. Perkas, R. Persky, A. Gedanken and D. Aurbach, Adv. Funct. Mater., 2010, 17, 1487–1492 CrossRef.
- M. S. Goh and M. Pumera, Anal. Bioanal. Chem., 2011, 399, 127–131 CrossRef CAS.
- T.-W. Chen, Z.-H. Sheng, K. Wang, F.-B. Wang and X.-H. Xia, Chem.–Asian J., 2011, 6, 1210–1216 CrossRef CAS.
- S. Guo, D. Wen, Y. Zhai, S. Dong and E. Wang, Biosens. Bioelectron., 2011, 26, 3475–3481 CrossRef CAS.
- Z. Naal, J. H. Park, S. Bernhard, J. P. Shapleigh, C. A. Batt and H. D. Abruna, Anal. Chem., 2002, 74, 140–148 CrossRef CAS.
- R. G. Smith, N. D'Souza and S. Nicklin, Analyst, 2008, 133, 571–584 RSC.
- A. Hilmi, J. H. T. Luong and A. L. Nguyen, J. Chromatogr., A, 1999, 844, 97–110 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2an36351h |
| This journal is © The Royal Society of Chemistry 2013 |