Integration of multiple components in polystyrene-based microfluidic devices part II: cellular analysis
Kari B.
Anderson
a,
Stephen T.
Halpin
a,
Alicia S.
Johnson
b,
R. Scott
Martin
*b and
Dana M.
Spence
*a
aDepartment of Chemistry, Michigan State University, 578 S. Shaw Blvd, East Lansing, Michigan, USA 48824. E-mail: dspence@chemistry.msu.edu; Fax: +1 517-353-1793; Tel: +1 517-355-9715x174
bDepartment of Chemistry, Saint Louis University, St. Louis, Missouri 63103, USA. E-mail: martinrs@slu.edu; Tel: +1 314-977-2836
First published on 19th October 2012
Abstract
In Part II of this series describing the use of polystyrene (PS) devices for microfluidic-based cellular assays: various cellular types and detection strategies are employed to determine three fundamental assays often associated with cells. Specifically, using either integrated electrochemical sensing or optical measurements with a standard multi-well plate reader, cellular uptake, production, or release of important cellular analytes are determined on a PS-based device. One experiment involved the fluorescence measurement of nitric oxide (NO) produced within an endothelial cell line following stimulation with ATP. The result was a four-fold increase in NO production (as compared to a control), with this receptor-based mechanism of NO production verifying the maintenance of cell receptors following immobilization onto the PS substrate. The ability to monitor cellular uptake was also demonstrated by optical determination of Ca2+ into endothelial cells following stimulation with the Ca2+ ionophore A20317. The result was a significant increase (42%) in the calcium uptake in the presence of the ionophore, as compared to a control (17%) (p < 0.05). Finally, the release of catecholamines from a dopaminergic cell line (PC 12 cells) was electrochemically monitored, with the electrodes being embedded into the PS-based device. The PC 12 cells had better adherence on the PS devices, as compared to use of PDMS. Potassium-stimulation resulted in the release of 114 ± 11 μM catecholamines, a significant increase (p < 0.05) over the release from cells that had been exposed to an inhibitor (reserpine, 20 ± 2 μM of catecholamines). The ability to successfully measure multiple analytes, generated in different means from various cells under investigation, suggests that PS may be a useful material for microfluidic device fabrication, especially considering the enhanced cell adhesion to PS, its enhanced rigidity/amenability to automation, and its ability to enable a wider range of analytes to be investigated, even analytes with a high degree of hydrophobicity.
A Introduction
Due to the ability to integrate multiple processes and fabricate structures that can easily mimic an in vivo environment, microfluidic devices have become popular as an enabling tool when studying cellular systems.1–4 PDMS is the most widely used substrate in microfluidic devices due to its transparent nature, ease of fabrication, low cost, and the fact that it is gas permeable.5,6 Although PDMS devices have been successfully used in a variety of applications for cellular analysis,1,7–9 there are some existing disadvantages of using PDMS including poor cell adhesion for some cell lines, partitioning of hydrophobic molecules in the PDMS, and leaching of un-crosslinked monomers from the bulk PDMS into the cell culture system.10Recently, several studies have focused on the use of thermoplastic-based microfluidic devices for on-chip cell culture because these substrates have been shown to be more biocompatible than PDMS.11–15 The most commonly used substrate in biological systems is polystyrene, the material from which most cell culture flasks are made, and there have been several recent reports of fabricating microfluidic devices in this material. Initially, Beebe's group developed simplified, embossing-based methods for fabricating such devices and demonstrated their utility by monitoring (via imaging) the upregulation of E-selectin in a monolayer of human umbilical vein endothelial cells that had been activated by interleukin 1β. Reports from other groups have included the fabrication of thin-layer polystyrene devices with micropallet arrays for improving cell adhesion and proliferation of primary muscle cells,13 as well as microwells for segregation and tracking of non-adherent and adherent cells.16 Midwoud et al. recently compared the use of different thermoplastics, including polystyrene, for the adherence of human hepatoma cells in a patterned structure, with the substrates being characterized in terms of surface treatment, adsorption of hydrophobic compounds, and biocompatibility.11 These studies have shown that polystyrene-based devices hold great promise for on-chip cell studies. However, there have not been examples of using polystyrene devices with integrated analytical functions to monitor intra- or extra-cellular function. For example, there has been a lack of integrating polystyrene devices with either on-chip processes (such as the use of electrodes for integrated detection) or existing research infrastructure used for high-throughput 96 well-plate studies (such as 8-channel pipets and plate readers).
The fabrication strategies for polystyrene devices, which were described in Part I of this series, are utilized to quantitate analyte production, release, or uptake in two different cell lines, namely, endothelial and PC 12 cells. The adhesion of PC 12 cells is compared on PDMS and native polystyrene surfaces that have been coated with collagen. Polystyrene devices with a PDMS injection block (for integrating a standard micropipette as part of the pumping mechanism) and serpentine microchannels (that can be integrated with a 96-well plate reader) were used to culture bovine pulmonary artery endothelial cells (bPAECs). Intracellular nitric oxide (NO) production and calcium (Ca2+) uptake in the endothelial cells were monitored in a high-throughput manner using fluorogenic probes and standard plate reader detection. Finally, it is shown that microchannels can be molded into a polystyrene device that also contains embedded electrodes. The resulting device with integrated electrodes was used to measure the stimulated release of catecholamines from PC 12 cells.
B Experimental
PC 12 culture on polystyrene devices
PC 12 cells (ATCC, Manassas, VA, USA) were cultured (at 37 °C and 6% CO2) in T-25 flasks (Fisher Scientific, Springfield, NJ, USA). The T-25 flasks and surfaces for cell adhesion were pre-treated with collagen solution, which was previously optimized to 0.435 mg mL−1.17 The PC 12 cells were grown to about 90% confluency with media (F-12K supplemented with 1% penicillin–streptomycin solution, 2.5% fetal bovine serum, and 15% horse serum, all from ATCC) being replaced every 1–2 days. When the cells reached confluency in the T-25 flasks (as determined by optical microscopy), the cells were either split into new flasks or used for on-chip experiments.For the cell adhesion study, a PDMS reservoir was sealed on either the PDMS or polystyrene surface and pre-treated with collagen. A cell suspension from a confluent T-25 flask was made by scraping the cells from the flask (in 5 mL of media), placing the solution in a 15 mL centrifuge tube (Dow Corning, Midland, MI, USA), and centrifuging at 110g for 3 minutes until a pellet of cells was formed. The supernatant was removed and fresh media was added. The pellet of cells was re-suspended in solution and 100 μL of the solution were added to the PDMS reservoir for the cell adhesion studies.
Endothelial cell culture in polystyrene-based microchannels
In order to facilitate cell adhesion in the channels of the polystyrene device, a 100 μg mL−1 solution of bovine plasma fibronectin (Invitrogen, Carlsbad, CA, USA) cell adhesion protein was introduced to the channels via the injection block and allowed to adsorb for 1 hour in a 37 °C and 5% CO2 incubator. The channels were then dried and exposed to UV light for 15 minutes. bPAECs were purchased as frozen cryovials (Lonza, Walkersville, MD, USA). The vials were thawed to room temperature and added to a T-75 tissue culture flask containing 9 mL of endothelial growth media (EGM) that had been equilibrated to 37 °C. The EGM consists of a low glucose (5.5 mM) Dulbecco's Modified Eagles Medium (DMEM, MIDSCI, St. Louis, MO, USA) supplemented with 2.5% v/v adult bovine serum (Sigma-Aldrich, St. Louis, MO, USA), 7.5% fetal bovine serum (Lonza, Walkersville, MD, USA), penicillin, streptomycin, and amphotericin B (MIDSCI, St. Louis, MO USA). bPAECs were allowed to grow in a humidified incubator at 37 °C and 5% CO2 until they were determined confluent by optical microscopy. Media was changed the day after plating and then every 2 days thereafter. The bPAECs were subcultured when the cells reached >80% confluence, as visualized by optical microscopy.In order to seed bPAECs into the channels of the microfluidic device, bPAECs were washed with 10 mL of HEPES and then treated with 5 mL of 0.25% trypsin–EDTA, which was then removed and the cells suspended in 10 mL of media. The cell suspension was removed from the flask and centrifuged at 1500g for 5 min. The supernatant was removed and the pellet was resuspended in 450 μL of equilibrated media. This concentrated cell solution was introduced to the channels in the same manner as the fibronectin and incubated for 1 hour at 37 °C and 5% CO2. After an hour of growth, the ECs were re-seeded. Media was subsequently changed every 2 hours. The ECs were allowed to grow to confluency overnight and used the day after seeding.
To verify cell confluency in the channels, 1 μM CMFDA cell tracker (Molecular Probes, Carlsbad, CA, USA) was pumped through the channels to fluorescently label the bPAECs for monitoring with an optical microscope (Olympus IX71 Microscope, Olympus America, Melville, NY, USA) fitted with a FITC filter cube (Chroma Technology Corp, Bellows Falls, VT, USA) containing the excitation (460–500 nm) and emission (505–560 nm) filters.
Fluorescence detection of intracellular endothelial nitric oxide
A 5 mM stock solution of the intracellular nitric oxide (NO) probe DAF-FM-DA (4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate, Molecular Probes, Carlsbad, CA, USA) was made by dissolving 50 μg in 20 μL anhydrous dimethyl sulfoxide (DMSO). A 10 μM solution of DAF-FM-DA was made by diluting 2 μL of stock to 1 mL with physiological salt solution (PSS). The concentrations of salts (in mM) in the PSS were as follows: 4.7 KCl, 2.0 CaCl2, 1.2 MgSO4, 140.5 NaCl, 21.0 Tris, and 5.5 glucose (pH 7.4). The 10 μM DAF-FM-DA was then pumped (via the PDMS injection block) over the bPAECs using a 100 μL pipet set to 40 μL, and then repeated to ensure adequate cell exposure to probe. The device was then incubated for 1 hour at 37 °C and 5% CO2. After incubation, the channels were rinsed twice with PSS to remove excess probe, followed by a waiting period of 10 minutes at 37 °C and 5% CO2 to allow for probe de-esterification by intracellular mechanisms.Initial fluorescence measurements of the bPAECs were taken by aligning a 96 well plate, with drilled out wells corresponding to each channel's serpentine, on top of the PS device. Fluorescence measurements were performed using a plate reader (Spectramax M4, Molecular Devices, Sunnyvale, CA, USA) set to an excitation wavelength of 495 nm and an emission wavelength of 521 nm. These measurements served to represent the baseline levels of NO in the cells. After measurement, adenosine triphosphate (ATP, Sigma-Aldrich, St. Louis, MO USA) was pumped over the bPAECs in the same manner as the probe. ATP standards were prepared by dissolving ATP in 25 mL PSS to make a 1 mM stock solution. Next, a 100 μM solution was prepared by diluting 100 uL of the ATP stock solution to 1 mL; this 100 μM solution was then diluted further to prepare a 0.5 μM ATP solution that was subsequently pumped over the bPAECs in the device. Following a 30 minute incubation at 37 °C and 5% CO2, the final fluorescence measurements were taken on the plate reader in the same manner as before. The differences in fluorescence were taken and normalized to the channels that had been addressed with 0 μM ATP (PSS alone, see Fig. 3B).
Fluorescence detection of endothelial calcium uptake
A 2.3 mM stock solution of the intracellular calcium (Ca2+) specific fluorescence probe Fluo-4 AM (Invitrogen, Grand Island, NY, USA) was made by dissolving 50 μg in 20 μL anhydrous DMSO. Next, a 5 μL aliquot of the stock solution, along with 5 μL 200 mg mL−1 pluronic F-127 surfactant (Invitrogen, Grand Island, NY, USA), were combined in order to enhance the probe's penetration capacity. Finally, the probe solution was diluted to 2 mL in Ca2+ free PSS to create a 5.7 μM solution. The 5.7 μM Fluo-4 AM was then pumped over the bPAECs using a 100 μL pipet set to 40 μL (via the PDMS injection block), then repeated to ensure adequate cell exposure to probe. The device was then incubated for 30 min at 37 °C and 5% CO2. After incubation, the channels were rinsed twice with Ca2+ free PSS to remove excess probe and incubated for an additional 10 minutes at 37 °C and 5% CO2 to allow for probe de-esterification by intracellular mechanisms. Initial fluorescence measurements of the bPAECs were obtained using the plate reader as described above for the NO, although the excitation and emission wavelength settings were modified to 496 nm and 516 nm, respectively. These measurements served to represent the baseline levels of Ca2+ in the cells.After measurement, PSS (with Ca2+) or 10 μM Ca2+ ionophore A23187 (Sigma-Aldrich, St. Louis, MO USA) in PSS were pumped over the bPAECs in the same manner as the probe. The Ca2+ ionophore was prepared by dissolving 5 mg in 1.9 mL anhydrous DMSO to create a 5 mM stock solution. Next, a 100 μM working solution was prepared by diluting 20 μL of stock to 1 mL in PSS and finally, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 dilution in PSS was performed, resulting in a final concentration of 10 μM Ca2+ ionophore. Once PSS or ionophore was flowed over the bPAECs, the device was allowed to incubate for 10 minutes at 37 °C and 5% CO2. Then final fluorescence measurements were taken on the plate reader as described above.
:10 dilution in PSS was performed, resulting in a final concentration of 10 μM Ca2+ ionophore. Once PSS or ionophore was flowed over the bPAECs, the device was allowed to incubate for 10 minutes at 37 °C and 5% CO2. Then final fluorescence measurements were taken on the plate reader as described above.
Fabrication and assembly of capillary-molded polystyrene channels with embedded electrodes
If desired, polystyrene microchannels can also be incorporated into polystyrene substrates that have embedded electrodes already shaped by wet polishing. These channels were formed by placing a capillary that has an outer diameter matching the desired channel width onto the polystyrene substrate. For these studies, fused-silica capillary (Polymicro Technologies, Phoenix, AZ, USA) tubing having a length of 1 cm and an outer diameter of 150 μm was used to define channel dimensions. A small piece of glass was used to hold the capillary in place while heating for about 20 s with a heat gun. After these 20 s, the glass was removed and the imprinting of the capillary was achieved with an additional 20 seconds of heating with the heat gun. The heat gun heats the polystyrene to 150 °C, a temperature that allowed for imprinting of the capillary into the PS surface. After cooling, the capillary was removed from the polystyrene substrate. The channel depth was measured using a profilometer (Dektak3 ST, Veeco Instruments, Woodbury, NY, USA). For these studies, the channels were 70 μm deep.For device assembly, a PDMS flow channel was sealed at the edge of the polystyrene channel to form an interface for coupling flow from the polystyrene channels with the embedded electrodes. Next, the PDMS channel was also sealed over a carbon fiber bundle detection electrode and a Pt counter electrode. A 20-gauge Luer stub adapter (Becton Dickinson, Sparks, MD, USA) was used to punch a hole at the end of the PDMS channel where fluidic tubing was inserted. A bi-directional syringe pump (Eldex MicroPro, Napa, CA, USA) set in withdraw mode was used to pull the sample through the channels at a flow rate of −1.5 μL min−1. In this manner, solution was constantly withdrawn from the reservoir, through the polystyrene channels, into the overlaid PDMS channels, and over the embedded electrodes.
Use of polystyrene substrates for PC 12 cell analysis
The final constructed device was utilized for analyzing the release of catecholamines from PC 12 cells. The polystyrene in the PDMS-defined reservoir was pre-treated with collagen. The contents of a confluent T-25 flask were split for on-chip experiments and 100 μL of the cell solution was placed in the reservoir. After plating of the cells, the device was incubated (at 37 °C and 6% CO2) for 2 hours. For reserpine inhibition studies, 100 μM reserpine (Sigma Aldrich, St. Louis, MO, USA) was incubated with the cells for 2 hours. After incubation, the cells were then rinsed with a cell-compatible buffer (4.2 mM KCl, 150 mM NaCl, 2 mM CaCl2, 0.7 mM MgCl2, 1 mM NaH2PO4, and 10 mM HEPES, Sigma Aldrich, St. Louis, MO, USA). The polystyrene and PDMS channels were allowed to fill with the cell buffer. The fluidic tubing was inserted and the syringe pump began to pull the cell buffer through the channels and over the electrodes. After equilibration with the cell buffer, a stimulant buffer (80 mM KCl, 50 mM NaCl, 2 mM CaCl2, 0.7 mM MgCl2, 1 mM NaH2PO4, and 10 mM HEPES) was introduced to the reservoir and pulled through the channels for 10 seconds. The reservoir contents were then changed back to the cell compatible buffer. Amperometric detection of the catecholamine release was achieved at +0.9 V vs. the on-chip Pt counter electrode. The non-fluorescent images were taken with a stereoscope (Olympus SZ61) operating in bright-field mode using a Sony 3CCD color camera (Leeds Precision Instruments, Minneapolis, MN, USA). The interface micrograph and the cell image were obtained using an upright microscope (Olympus EZ 60 equipped with a Qicam Fast digital CCD camera) operating in bright-field mode.C Results and discussion
PC 12 cell adhesion studies
A limitation of PDMS devices is poor cell adhesion of some cell lines, such as PC 12 cells. Adherent proteins such as collagen or fibronectin can be coated onto PDMS surfaces, but many cells are still not as adherent on PDMS as they are in conventional cell culture substrates such as polystyrene. Previous studies have investigated the biocompatibility of cells on various thermoplastics including polystyrene, with the surfaces being rendered hydrophilic by means of UV-ozone or plasma treatment.11,16 In this study, PC 12 cell adhesion was investigated on either a native polystyrene device that was fabricated from polystyrene powder (as described in Part I of this series) or a PDMS device. In both cases, the substrates were pre-treated with collagen. A cell suspension was made from a 90% confluent T-25 flask of PC 12 cells and a 100 μL aliquot of the suspension was added to a reservoir (defined by PDMS) that was sealed over either a PDMS or polystyrene surface. The cells were incubated on the polystyrene and PDMS for 2 hours. After 2 hours, the substrates were removed from the incubator for inspection. As seen in Fig. 1, the PC 12 cells were much more adherent on the polystyrene surface, especially after the cells were rinsed with media. In duplicate studies (one of which is shown in Fig. 1) it was found that after 3 rinses with cell media, 75% of the cells were lost from PDMS surface, while only 8% of the cells were detached from the polystyrene surface after rinsing. This study clearly shows that polystyrene-based microfluidic devices are preferable over PDMS-devices for analysis of adherent cells. Of course, most non-viable cells will detach when exposed to flow. Therefore, an important aspect of cell-based assays using microfluidic technologies is ensuring that cells are adherent when subjected to flow; this characteristic is significantly enhanced when using the PS-based devices.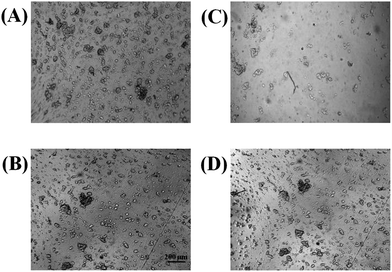 | ||
| Fig. 1 Bright field micrographs demonstrating PC 12 cell adhesion to PDMS (A) and polystyrene (B). As expected, both substrates provide a sufficient surface for cell adhesion. However, after washing 3× with buffer, the number of cells adhering to PDMS (C) is reduced in comparison to cells adhering to polystyrene (D). | ||
Monitoring nitric oxide production and calcium uptake in endothelial cells
The pumping mechanism using the PDMS injection block to interface with a pipet (as described in Part I of this work) was used to introduce bPAECs into to the polystyrene-based microchannels. A side view of one channel is shown in Fig. 2, where a layer of fibronectin (cell adhesion protein) was first introduced to the channel (via the injection block). After allowing the fibronectin to absorb for 1 hour, a concentrated solution of bPAECs was introduced into the device and allowed to adhere and grow in the microchannel (in an incubator), with frequent media changes (every 2 hours). The bPAECs on the polystyrene grew to confluence overnight and were visualized by optical microscopy as shown in the bright field image in Fig. 2. To further ensure that the cells in the serpentine channels were confluent, they were also labeled with the cell tracker CMFDA and visualized under the FITC filter of the microscope (as pictured in the fluorescent image, Fig. 2). Before any biological studies were performed on the bPAECs cultured in the polystyrene device, they were first visualized with optical microscopy to confirm the presence of healthy, confluent cells (Fig. 3A).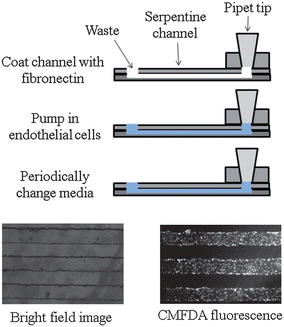 | ||
| Fig. 2 Endothelial cell culture on a polystyrene device. To culture endothelial cells, the device is first coated with fibronectin. Next, a suspension of endothelial cells is pumped into the device. The device is then incubated at 37 °C, 5% CO2 for 2 hours. Media is changed every two hours until cell confluence is observed. A bright field image is shown on the left, while the image on the bottom right was obtained by incubating the cells with CellTracker CMFDA fluorescence. | ||
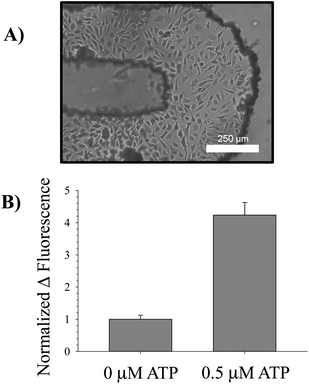 | ||
| Fig. 3 Intracellular measurement of NO. A bright field micrograph of endothelial cells cultured in the turn of a serpentine channel is provided in (A). These cells were incubated with 10 μM DAF-FM-DA, an intracellular NO probe, at 37 °C for 30 min; excess probe was rinsed off the cells by pumping buffer without probe through the channels. After the incubation period, a basal fluorescence measurement was taken using a standard plate reader. Next, 0.5 μM ATP was pumped over the cells and allowed to incubate for an additional 30 min in order to stimulate NO production in the endothelial cells. A final fluorescence measurement was taken using the plate reader and the difference in fluorescence calculated. This value was normalized to the difference in fluorescence for a 0 μM ATP standard. The average changes in fluorescence for n = 3 separate devices is shown in (B), along with error bars representing the standard error of the mean. The changes are significant for p < 0.001. | ||
The in-channel cultured bPAECs were incubated with the intracellular NO probe DAF-FM-DA and then exposed to ATP, a known stimulus of NO production in endothelial cells;7,18 therefore, it was anticipated that ATP would result in increased intracellular bPAEC NO production. Upon application of 0.5 μM ATP to the cells in the serpentine channel, a significant increase in NO production was observed when compared to applying PSS alone (Fig. 3B). This increase was measured by aligning the device in the plate reader and performing top-read fluorescence measurements in a high-throughput manner. The device was also utilized to measure cellular uptake by monitoring Ca2+ influx into the bPAECs after stimulation by a Ca2+ ionophore. Cells in the device treated with Ca2+ ionophore exhibited a 42 ± 13% increase in fluorescence compared to cells treated with PSS alone, which only exhibited a 17 ± 2% increase in fluorescence (N = 3, error: SEM, p-value < 0.05). These studies suggest that the polystyrene devices with imprinted channels can be integrated with existing infrastructure (pipets for pumping and plate readers for detection) and can be utilized to study both an intracellular process (NO production) and cellular uptake (Ca2+ influx).
Capillary-based molding of polystyrene channels for integration of cell culture with electrochemical detection
An innovative feature of the polystyrene devices is that, in addition to fabricating microchannels, polystyrene-embedded electrodes can also be integrated. Polystyrene-embedded electrodes were fabricated as described in Part I of these manuscripts. It was found that if PDMS-based structures were used to imprint channels in the polystyrene (via heating on a hotplate), the resulting polystyrene device surface became uneven with some bubble formation, especially around the embedded electrodes. To address this, a fused-silica capillary of desired length and diameter (1 cm long, 150 μm o.d.) was placed on the polystyrene substrate and covered with a small piece of glass. A heat gun was utilized to heat the glass and capillary for 20 seconds (Fig. 4A). The glass was then removed and the capillary was heated for an additional 20 seconds (Fig. 4B). The glass transition temperature of PS is approximately 95 to 100 °C.12 The heat gun heats the polystyrene surface to ∼150 °C (as measured by a thermocouple) to enable imprinting of the capillary into the PS surface. The substrate and capillary were then cooled to room temperature before removal of the capillary, resulting in polystyrene channel depths of 70 μm. The localized heating nature of this approach did not result in any issues with deforming the polished polystyrene substrate or the embedded electrodes.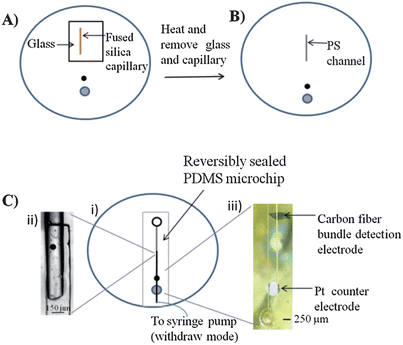 | ||
| Fig. 4 Fabrication and assembly of polystyrene microchannels integrated with embedded electrodes. (A) A 150 μm o.d. capillary is placed on a polystyrene base with embedded electrodes. A small glass plate is used to hold the capillary in place. The capillary is heated for 20 seconds with a heat gun before the glass plate is removed. (B) The capillary is further heated another 20 seconds before the capillary is removed and a polystyrene channel remains. (C) (i) Top down view of the assembled device. (ii) A PDMS flow channel is sealed at the interface of the polystyrene channel. (iii) Micrograph of the PDMS flow channel sealed over the carbon fiber bundle detection electrode and the Pt counter electrode. | ||
The procedure depicted in Fig. 4C, i was utilized to assemble a device for integrating the embedded electrodes with the imprinted channels. A key feature of this procedure is the overlay of the imprinted channel with a PDMS-based microchannel (Fig. 4C, ii). The PDMS microchannel was used to interface the fluidic structure with the embedded working and counter electrodes (Fig. 4C, iii). By using a bi-directional pump set to withdraw mode, fluid was continuously pumped from the reservoir, through the polystyrene channel to the PDMS flow channel interface, and over the embedded electrode surface.
The device shown in Fig. 4C was used for the cell studies. PC 12 cells were immobilized in the reservoir leading to the imprinted channels. PC 12 cells (from a 90% confluent T-25 flask) were plated onto the PS surface and imprinted channels and incubated for 2 hours. A micrograph of the reservoir with a confluent layer of PC 12 cells is shown in Fig. 5A. The fluidic tubing was inserted into the microchip and the bi-directional syringe pump was set on withdraw mode at a flow rate of −1.5 μL min−1, exposing the cells to a cell-compatible buffer. After a steady state current was achieved, the stimulated release of catecholamines (dopamine + norepinephrine) from the PC 12 cells was initiated by replacing the cell buffer with a K+ stimulant solution19 for 10 seconds, after which time the stimulant solution was removed and replaced with cell-compatible buffer. The introduction of the stimulant resulted in catecholamine release that was subsequently detected at the carbon fiber bundle detection electrode (Fig. 5B). Reserpine is a known inhibitor of the vesicular monoamine transporter and inhibits neurotransmitter release by displacing catecholamines from neurotransmitter vesicles.19 For inhibition studies, the on-chip plated PC 12 cells were incubated with 100 μM reserpine for 2 hours, followed by addition of the K+ stimulant solution as described above. The amperograms resulting from a 100 μM dopamine standard, the stimulated release of catecholamines, and the reserpine-inhibition of the cells are shown in Fig. 5B. Calibration was performed using 100 μM injections of a dopamine standard thereby enabling quantitation of catecholamine release. Such a determination was performed using 3 different chips on different days (with similar cell confluency). As shown in Fig. 5C, the average K+ stimulated release for the untreated PC 12 cells was 114 ± 11 μM, while the stimulated release for cells that had been inhibited with reserpine was 20 ± 2 μM. It was also found that operating the syringe pump in withdraw mode to pull buffer/stimulant over the cells and through the fluidic network, as opposed to our previous studies where buffer/stimulant was pumped over the cells with a positive displacement,9,17 led to no issues with air bubbles or cells becoming detached from the surface over time. This study shows that the versatile nature of the fabrication strategies developed in both Parts I and II of this work enables not only monitoring of intracellular and uptake processes (previous section), but also extracellular release with integrated channels and electrodes.
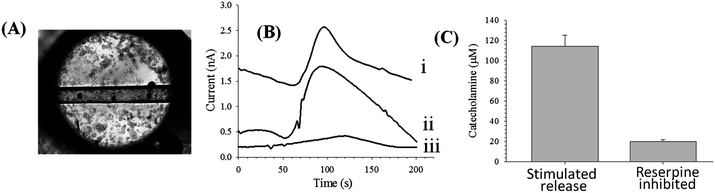 | ||
| Fig. 5 Use of polystyrene substrates for PC 12 cell analysis. (A) Micrograph of PC 12 cells on polystyrene surface as well as in a polystyrene microchannel. (B) Amperograms of 100 μM dopamine (i), K+ stimulated release (ii), and reserpine-inhibited release (iii). (C) Quantitative comparison of K+ stimulated release and reserpine-inhibited release (for 3 different chips on different days with a similar cell confluence). | ||
D Conclusions
The fabrication strategies for polystyrene devices that were explained in Part I of this series were employed to investigate different cellular processes using two completely different types of cell lines. First, endothelial cells, cells that typically line the inner wall of blood vessels, were immobilized into a polystyrene device. These cells were stimulated to produce nitric oxide, which was determined intracellularly with an optical fluorescence probe. Furthermore, these cells' ability to uptake Ca2+ was also determined with an intracellular fluorescence probe. In a separate set of experiments, the stimulated (K+) release of catecholamines from a layer of immobilized PC 12 cells was performed on a polystyrene device, with inhibition studies also being carried out. Importantly, the electrodes (both working and counter) were embedded in the polystyrene and integrated with polystyrene-based microchannels onto which the cells were immobilized. These results demonstrate that devices fabricated in polystyrene are capable of being used in many of the same types of assays as those devices made from other common materials such as PDMS.Importantly, there are advantages associated with polystyrene in comparison to PDMS. First, it is shown here that collagen-coated polystyrene was a more suitable substrate for PC 12 cell adhesion when compared to PDMS. Furthermore, the polystyrene devices (containing a PDMS injection block and plate reader fluorescence detection capabilities) were successfully used to study NO production and Ca2+ uptake by ECs immobilized in the serpentine channels in a high-throughput manner. While previous work by the authors has integrated PDMS-based devices with plate readers, a more rigid material such as polystyrene will lend itself to further automation such as plate reader handlers that involve robotic arms and transit belts that move plates from one locale to another. A final advantage, shown in Part I of this two-part manuscript series, is that the polystyrene-based devices enable the end-user to perform assays on compounds with a higher degree of hydrophobicity. We anticipate that the use of polystyrene will become more widespread as the use of microfluidic devices for biologically based assays continues to increase, especially as the level of complexity of new assays (multi-organ analyses, “body-on-a-chip” applications, etc.) continue to increase.
References
- A. L. Bowen and R. S. Martin, Electrophoresis, 2010, 31, 2534–2540 CrossRef CAS.
- J. El-Ali, P. K. Sorger and K. F. Jensen, Nature, 2006, 442, 403–411 CrossRef CAS.
- R. S. Martin, P. D. Root and D. M. Spence, Analyst, 2006, 131, 1197–1206 RSC.
- D. M. Spence, N. J. Torrence, M. L. Kovarik and R. S. Martin, Analyst, 2004, 129, 995–1000 RSC.
- D. C. Duffy, J. C. McDonald, O. J. A. Schueller and G. M. Whitesides, Anal. Chem., 1998, 70, 4974–4984 CrossRef CAS.
- M. L. Kovarik, P. C. Gach, D. M. Ornoff, Y. Wang, J. Balowski, L. Farrag and N. L. Allbritton, Anal. Chem., 2012, 84, 516–540 CrossRef CAS.
- L. I. Genes, N. V. Tolan, M. K. Hulvey, R. S. Martin and D. M. Spence, Lab Chip, 2007, 7, 1256–1259 RSC.
- S. T. Halpin and D. M. Spence, Anal. Chem., 2010, 82, 7492–7497 CrossRef CAS.
- M. W. Li and R. S. Martin, Analyst, 2008, 133, 1358–1366 RSC.
- K. J. Regehr, M. Domenech, J. T. Koepsel, K. C. Carver, S. E. Ellison-Zelski, W. L. Murphy, L. A. Schuler, E. T. Alarid and D. J. Beebe, Lab Chip, 2009, 9, 2132–2139 RSC.
- P. M. van Midwoud, A. Janse, M. T. Merema, G. M. M. Groothuis and E. Verpoorte, Anal. Chem., 2012, 84, 3938–3944 CrossRef CAS.
- E. W. Young, E. Berthier, D. J. Guckenberger, E. Sackmann, C. Lamers, I. Meyvantsson, A. Huttenlocher and D. J. Beebe, Anal. Chem., 2011, 83, 1408–1417 CrossRef CAS.
- D. A. Detwiler, N. C. Dobes, C. E. Sims, J. N. Kornegay and N. L. Allbritton, Anal. Bioanal. Chem., 2012, 402, 1083–1091 CrossRef CAS.
- G. Mehta, J. Lee, W. Cha, Y.-C. Tung, J. J. Linderman and S. Takayama, Anal. Chem., 2009, 81, 3714–3722 CrossRef CAS.
- A. Chen, D. K. Lieu, L. Freschauf, V. Lew, H. Sharma, J. Wang, D. Nguyen, I. Karakikes, R. J. Hajjar, A. Gopinathan, E. Botvinick, C. C. Fowlkes, R. A. Li and M. Khine, Adv. Mater., 2011, 23, 5785–5791 CrossRef CAS.
- Y. Wang, J. Balowski, C. Phillips, R. Phillips, C. E. Sims and N. L. Allbritton, Lab Chip, 2011, 11, 3089–3097 RSC.
- M. W. Li, D. M. Spence and R. S. Martin, Electroanalysis, 2005, 17, 1171–1180 CrossRef CAS.
- R. S. Sprague, M. L. Ellsworth, A. H. Stephenson and A. J. Lonigro, Am. J. Physiol., 1996, 271, H2717–H2722 CAS.
- K. D. Kozminski, D. A. Gutman, V. Davila, D. Sulzer and A. G. Ewing, Anal. Chem., 1998, 70, 3123–3130 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |