Positively charged polymer brush-functionalized filter paper for DNA sequence determination following Dot blot hybridization employing a pyrrolidinyl peptide nucleic acid probe†
Praethong S.
Laopa
ab,
Tirayut
Vilaivan
c and
Voravee P.
Hoven
*c
aProgram in Petrochemistry, Faculty of Science, Chulalongkorn University, Phayathai Road, Patumwan, Bangkok 10330, Thailand
bCenter for Petroleum, Petrochemicals, and Advanced Materials, Chulalongkorn University, Phayathai Road, Patumwan, Bangkok 10330, Thailand
cOrganic Synthesis Research Unit, Department of Chemistry, Faculty of Science, Chulalongkorn University, Phayathai Road, Patumwan, Bangkok 10330, Thailand. E-mail: vipavee.p@chula.ac.th; Fax: +66-2218-7598; Tel: +66-2218-7627 ext. 102
First published on 19th October 2012
Abstract
As inspired by the Dot blot analysis, a well known technique in molecular biology and genetics for detecting biomolecules, a new paper-based platform for colorimetric detection of specific DNA sequences employing peptide nucleic acid (PNA) as a probe has been developed. In this particular study, a pyrrolidinyl PNA bearing a conformationally rigid D-prolyl-2-aminocyclopentanecarboxylic acid backbone (acpcPNA) was used as a probe. The filter paper was modified to be positively charged with grafted polymer brushes of quaternized poly(dimethylamino)ethyl methacrylate (QPDMAEMA) prepared by surface-initiated polymerization of 2-(dimethylamino)ethyl methacrylate from the filter paper via ARGET ATRP followed by quaternization with methyl iodide. Following the Dot blot format, a DNA target was first immobilized via electrostatic interactions between the positive charges of the QPDMAEMA brushes and negative charges of the phosphate backbone of DNA. Upon hybridization with the biotinylated pyrrolidinyl peptide nucleic acid (b-PNA) probe, the immobilized DNA can be detected by naked eye observation of the yellow product generated by the enzymatic reaction employing HRP-labeled streptavidin. It has been demonstrated that this newly developed assay was capable of discriminating between complementary and single base mismatch targets at a detection limit of at least 10 fmol. In addition, the QPDMAEMA-grafted filter paper exhibited a superior performance to the commercial membranes, namely Nylon 66 and nitrocellulose.
Introduction
DNA sequence determination plays an important role in many biotechnology-related applications ranging from medical,1,2 forensic,3,4 agriculture and food sciences.5 Several methodologies have been continuously developed in order to attain practical and reliable techniques that should be simple, rapid, inexpensive, highly specific and sensitive. In addition, the ability to be implemented in a high-throughput fashion is a highly desirable feature from the economical perspective. Conventional detection mostly relies on the specific hybridization between the designed nucleic acid probe and the complementary sequence which is a part of the DNA analyte. The recognition event can then be transformed into a physically measurable variable which can be determined either by the techniques requiring labels/tags or by label-free techniques based on electrochemical,6–11 optical,12–14 and piezoelectric methods.5,15Since 1991, the Nielsen group has introduced a unique DNA analogue called peptide nucleic acid (PNA).16,17 In PNA, the negatively charged sugar-phosphate backbone of DNA is totally replaced by an uncharged peptide-like backbone (Scheme 1b). The reduced electrostatic repulsion between PNA and DNA backbones resulted in superior hybridization characteristics, e.g., higher thermal stability, stronger affinity with less dependence on the salt concentration, greater sequence specificity, and higher capability of strand invasion to double-stranded DNA. The high specificity of PNA makes it an excellent probe for DNA sequence determination. Despite these advantages, there are many attempts to improve the original PNA further during the past 20 years. Among several variants of PNA developed to date, the conformationally rigid pyrrolidinyl PNA derived from D-prolyl-2-aminocyclopentane carboxylic acid (acpc) backbones (acpcPNA),18,19 introduced by Vilaivan and co-workers (Scheme 1c), shows great promise because it can form a PNA·DNA duplex with even higher affinity and specificity than the original Nielsen's aegPNA. The potential of acpcPNA as a probe to detect the DNA base sequence has been recently demonstrated by employing various techniques including quartz crystal microbalance (QCM),20 surface plasmon resonance (SPR)21–23 and MALDI-TOF mass spectrometry.24 However, these techniques require advanced instruments that are suitable for only well-equipped laboratories and hence cannot be used on-site in remote or rural areas having limited clinical experts and medical facilities.
 | ||
| Scheme 1 Structures of (a) DNA, (b) Nielsen's aegPNA, and (c) Vilaivan's acpcPNA. | ||
Dot blot hybridization is a technique widely used in molecular biology and genetics for detecting biomolecules. The sample containing the biological target (mostly proteins or DNA) to be detected is spotted directly on a membrane (such as nitrocellulose, Nylon 66 and poly(vinylidene fluoride) (PVDF)) without prior separation. The presence or absence of a specific target can be detected by binding with a probe that can report the binding event by radioactivity,25 fluorescence,26 chemiluminescence,27–29 or enzyme-based colorimetric assays.26,30–35 Dot blot hybridization can analyze multiple samples inexpensively with high accuracy and in a high-throughput fashion.
In order to develop an assay based on the concept of Dot blot hybridization, three major components must be considered including the membrane, the probe and the detection method. Commercial membranes designed for DNA blotting including nitrocellulose,26,33 PVDF,36 and Nylon 6637 are generally hydrophobic, which may cause undesirable non-specific adsorption that complicates the analysis. This unwanted characteristic becomes a serious problem particularly with hydrophobic probes such as PNA. To suppress such non-specific interactions, a strong buffer solution containing metal ion (MgCl2),32 organic solvent (acetonitrile, formamide),24,32,33,37 anionic detergent (sodium dodecyl sulfate)24,32,33,37 or combinations thereof must be employed, which may negatively affect the PNA·DNA hybridization. Previously, we have succeeded in using Q-sepharose as an anion-exchanger for capturing the acpcPNA·DNA hybrid in combination with mass spectrometry as a platform for multiplex SNP detection in DNA samples.24 Based on a similar principle, this research aims to develop a hydrophilic and positively charged membrane for DNA sequence analysis in Dot blot format using acpcPNA probes. It is anticipated that the positive charges on the membrane not only provide a platform for selective capturing of PNA·DNA hybrids, but should also help preventing non-specific adsorption of other non-DNA components in the samples as well as the PNA probe itself.
Due to its availability and inexpensiveness, filter paper-based platforms for analytical applications have received considerable attention in recent years.38,39 In this research, a hydrophilic and positively charged membrane is developed from filter paper functionalized with quaternized poly(2-(dimethylamino)ethyl methacrylate) (QPDMAEMA) brushes.40–42 Although the formation of the surface-grafted polymer brushes of QPDMAEMA on a cellulosic membrane has been previously described,40 to the best of our knowledge, their specific application in DNA sequence determination has never been reported. In principle, the QPDMAEMA-grafted filter paper should readily capture the negatively charged DNA, but not the neutral acpcPNA, by electrostatic interaction. Only when the sequences of the DNA and the biotinylated acpcPNA probe (b-PNA) are complementary, the probe will be immobilized through hybridization with the surface-bound DNA. The non-complementary as well as the excess PNA probes may be removed by simple washing. The presence of the b-PNA·DNA hybrid on the membrane can then be visualized by an enzyme-based colorimetric assay employing horseradish peroxidase streptavidin (SA–HRP) conjugate and a chromogenic substrate such as o-phenylenediamine (OPD)26,30–35 (Fig. 1).
 | ||
| Fig. 1 Schematic representation of enzymatic amplified colorimetric detection of DNA following Dot blot hybridization using filter paper functionalized with positively charged polymer brushes and the peptide nucleic acid probe. | ||
Experimental section
Materials
Whatman no. 1 filter paper was used as the membrane. 2-(Dimethylamino)ethyl methacrylate (DMAEMA) (Aldrich, 98%) was passed through a column filled with basic alumina to remove the inhibitor. The ligand, tris[2-(dimethylamino)ethyl]amine (Me6TREN), was synthesized according to the published procedure43 from tris(2-aminoethyl)amine (98%, Aldrich). Ethyl 2-bromoisobutyrate (EBiB) (Fluka, >97%), 2-bromoisobutyryl bromide (Aldrich, 98%), copper(II) bromide (Fluka, 99%), tin(II) 2-ethylhexanoate (Sn(EH)2) (Aldrich, 95%), L-ascorbic acid (Sigma-Aldrich, >99%), 4-N,N-dimethylaminopyridine (DMAP) (Fluka, 95%), methyl iodide (Aldrich, >99%), streptavidin–horseradish peroxidase conjugate (SA–HRP) (Sigma-Aldrich, ≥80% protein content, 80–150 units per mg protein), bovine serum albumin (BSA) (Sigma-Aldrich, >96%), o-phenylenediamine (OPD) (Aldrich, 98%), 3,3′,5,5′-tetramethylbenzidine (TMB) (Sigma-Aldrich, ≥99%), urea–hydrogen peroxide (urea–H2O2) substrate kit (Merck, for analysis 35%), blotting-Nylon 66 membranes, type B, positive (Fluka, 0.45 μm pore size), Hybond ECL nitrocellulose membrane or Amersham Hybond™ ECL™ (GE Healthcare Life Sciences, 0.45 μm pore size) were used as received. All solvents used were of reagent grade. Oligonucleotides were purchased from Bioservice Unit, National Science and Technology Development Agency (Thailand). Ultrapure distilled water was obtained after purification using a Millipore Milli-Q system (USA) that involves reverse osmosis, ion exchange, and filtration steps.Instrumentation
1H and 13C NMR spectra were recorded in a solution of CDCl3 or DMSO-d6 on a Varian Mercury-400 nuclear magnetic resonance spectrometer operating at 400 MHz. Chemical shifts were reported in parts per million (ppm) relative to tetramethylsilane (TMS). Infrared spectra of materials scraped from the surface-modified filter paper and prepared as KBr pellets were collected on a Nicolet Impact 6700 FT-IR spectrometer with 32 scans at a resolution of 4 cm−1 in a frequency range of 400–4000 cm−1. The molecular weight and polydispersity index of the free poly(2-(dimethylamino)ethyl methacrylate (PDMAEMA) formed in solution were measured using a Waters GPC system (USA), performed at 35 °C using THF (1.0 mL min−1) as the mobile phase and a Waters E600 column connected to the RI detector. Narrow polystyrene standards were used for generating a calibration curve. The water contact angles were measured in air at ambient temperature using a contact angle goniometer, model 200-F1, equipped with a Gilmont syringe and a 24-gauge flat-tipped needle (Ramé-Hart, USA). The data for each sample were taken from five different areas of the substrate and analyzed by DROPimage standard 2.0, after which they were expressed as the arithmetic mean value ± standard deviation (SD). The surface morphology of the filter paper before and after stepwise modification was observed by SEM on a JEOL (JSM-6480LV, Japan) instrument. Surface composition of the surface-modified filter paper was characterized by X-ray photoelectron spectroscopy (XPS) on a Physical Electronics Quantum 2000, using monochromatic Al k-alpha X-rays, and standard dual ion-electron neutralization.The mass spectra of acpcPNA were recorded on a Microflex MALDI-TOF mass spectrometer (Bruker Daltonics, Germany). The sample (1 μL) was mixed with 10 μL of the matrix solution consisting of α-cyano-4-hydroxycinnamic acid (CCA) in 0.1% TFA in acetonitrile–water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solution. This mixture (1 μL) was deposited onto the target, allowed to dry, and analyzed in positive ion linear time-of-fight mode with an accelerating voltage of +20 kV. All spectra were recorded by averaging between 20 and 30 individual laser shots.
:1) solution. This mixture (1 μL) was deposited onto the target, allowed to dry, and analyzed in positive ion linear time-of-fight mode with an accelerating voltage of +20 kV. All spectra were recorded by averaging between 20 and 30 individual laser shots.
The scanned images of the tested results on filter papers were recorded on a XEROX Workcentre 3119 scanner in 24 bit RGB mode. The brightness/contrast/resolution was set to 128/128/300. The images were saved as TIFF-files. The intensity of each spot was determined using Scion Image software by first converting to gray scale at 300 dpi. Intensity measurements were carried out using the Line tool to select an area for analysis to obtain profile images.
Preparation of QPDMAEMA-grafted filter paper
The initiator immobilized filter paper was prepared according to the published procedure by Lee and co-workers.40 The filter paper (3.5 × 8 cm2) was first cleaned by sonicating in dichloromethane (CH2Cl2) for 2 min prior to immersion in a solution of triethylamine (2.4 mL, 17 mmol) and a catalytic amount of DMAP (39 mg, 0.3 mmol) in 25 mL of CH2Cl2 under stirring for 10 min. Then, 2-bromoisobutyryl bromide (0.6 mL, 1.9 mmol) was slowly added to this solution. After the reaction proceeded for 16 h at ambient temperature (27–33 °C) with gentle agitation, the filter paper was thoroughly rinsed with CH2Cl2 followed by methanol and then air-dried.The initiator immobilized filter paper was placed in a 32 mL scintillation vial containing DMAEMA (16.8 mL, 100 mmol), CuBr2 (1.2 mg, 0.005 mmol), Me6TREN (57.0 mg (0.25 mmol), and EBiB (74 μL, 0.5 mmol) dissolved in 4.5 mL acetone. After stirring for 10 min, a solution of Sn(EH)2 (490 mg, 1.2 mmol) in 3 mL acetone was then added into the mixture. The vial was then sealed with a rubber septum. The volume of free space above the solution was 7.7 mL. The reaction was left stirring for a specified reaction time (24 h) at ambient temperature (27–33 °C). The filter paper was removed from the vial, successively washed with THF and MeOH to remove the residual monomer and catalysts, and was finally air-dried to yield PDMAEMA-grafted filter paper.
The PDMAEMA-grafted filter paper was placed in 25 mL DMF containing an excess amount of methyl iodide (100 μL). After stirring for 20 h, the filter paper was removed from the solution and washed three times with MeOH followed by THF under sonication and then air-dried. The filter paper was immersed in 100 mL of 0.1 M NaCl solution for 24 h, soaked in 500 mL of deionized water for 48 h and was finally air-dried to obtain QPDMAEMA-grafted filter paper.
Preparation of the biotinylated acpcPNA probe
Three acpcPNA probes, two 13 mer sequences (SLE1 and SLE2) corresponding to two different SNPs at the position −1082 of human IL-10 promoter gene and one 12 mer random sequence (TG), were used in the study (Table 1). We chose PNA probes with length between 12 and 13 bases which should give a good balance between sensitivity and specificity. The practicality of such 13-base long probes for detection of real human DNA samples has previously been demonstrated.24 The biotinylated acpcPNA probes (b-PNA) were synthesized according to literature procedures.18–24 The biotin was attached at the N-termini of acpcPNA while still on the solid support via two successive aminoethoxyethoxyacetyl (egl) linkers employing biotin pentafluorophenyl ester in the presence of HOAt/DIPEA. After nucleobase side-chain deprotection and cleavage from the solid support, the crude b-PNA was purified by reversed-phase HPLC with UV detection at 260 nm. A Varian Polaris C18 analytical HPLC column (3 μm particle size 4.6 × 50 mm) was used and eluted with a gradient of 0.1% TFA in MeOH and 0.1% TFA in water. After purification, the solution of PNA was lyophilized and the identity of the acpcPNA oligomers was confirmed by MALDI-TOF mass spectrometry and by thermal denaturation experiments (after hybridization with complementary DNA sequences) (Table 1).| Name | PNA sequence | m/z (calculated) | m/z (observed) | T m (°C) |
|---|---|---|---|---|
| b-PNA (SLE1) |
N-Biotin-(egl)2-TTCCCC![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) TCCCAA-LysNH2-C TCCCAA-LysNH2-C |
4880.36 | 4879.92 | 59.3 |
| b-PNA (SLE2) |
N-Biotin-(egl)2-TTCCCC![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) TCCCAA-LysNH2-C TCCCAA-LysNH2-C |
4895.37 | 4898.64 | 65.4 |
| b-PNA (TG) | N-Biotin-(egl)2-TGATGCTATGAC-LysNH2-C | 4722.12 | 4724.46 | not measured |
General protocol for DNA sequence determination
The QPDMAEMA-grafted filter paper was cut into 1.0 × 7.0 cm2 pieces and the spot positions were marked with a pencil. The designated DNA and b-PNA sequences from the list in Table S1 (ESI†) were spotted on the filter paper following a sequence indicated in Table S2 (ESI†). Two microliters of the DNA sample (1 μM in the solution of 0.4 M NaOH containing 10 mM EDTA) was spotted on the filter paper using a micropipette. The filter paper was air-dried and 2 μL of the b-PNA probe (1 μM solution in 0.1 M sodium phosphate buffer (PBS) pH 7.4) was subsequently introduced. The filter paper was rinsed immediately three times with phosphate buffer solution and Milli-Q water followed by incubation in a blocking solution (1% BSA, w/v) at ambient temperature for 30 min. Two microliters of SA–HRP (20 μg mL−1) was then spotted onto the filter paper at the position where the DNA sample was immobilized. The filter paper was then rinsed three times with PBS, Milli-Q water, and citrate buffer (CTB, 0.1 M, pH 5). Two concentrations of the PBS solutions (0.1 M or 50 mM) with or without added 0.1 M NaCl used in the step of washing and blocking were tested in order to find the optimal condition that yields the most efficient detection. Without drying, the filter paper was immersed in a solution consisting of 250 μL of 1.6 mg mL−1 OPD substrate (freshly prepared prior to use) and 250 μL of 1.6 mg mL−1 urea–H2O2 for 1 min and was finally washed with deionized water.Results and discussion
Preparation and characterization of QPDMAEMA-grafted filter paper
The QPDMAEMA-grafted filter paper was prepared by surface-initiated polymerization (SIP) of 2-(dimethylamino)ethyl methacrylate (DMAEMA) on the filter paper followed by quaternization with methyl iodide (Scheme 2). The initiator immobilized filter paper was prepared by acylation of the hydroxyl groups on the filter paper with 2-bromoisobutyryl bromide in the presence of triethylamine/DMAP. The surface of the filter paper became hydrophobic with a water contact angle of 125.5 ± 1.0° (image of the water droplet is shown in Fig. S1a, ESI†), as opposed to the completely wettable virgin filter paper (water contact angle ∼ 0°) suggesting that the immobilization of the 2-bromoisobutyrate moieties on the filter paper was successful. This water contact is much greater than that observed on a gold-coated glass slide grafted onto a monolayer of 2-bromoisobutyrate groups (72°).44 It is suspected that the additional hydrophobicity may come from the intrinsic roughness of the filter paper causing the water drop to be pinned while performing contact angle measurement. It should be emphasized that this unexpected high contact angle was only observed when at least 16 h of reaction time was used. In the presence of a catalytic amount of DMAP, it is believed that acylation should be more effective and thus yielded a greater graft density of 2-bromoisobutyrate groups on the filter paper than that of the work reported previously by Carlmark and Malmstrom45 of which the acylation was conducted in the absence of a catalyst. Intuitively, the wetting behavior of the filter paper that may be less covered by 2-bromoisobutyrate groups should be largely governed by the extremely hydrophilic nature of the origin filter paper. This may be the reason why the contact angle of their 2-bromoisobutyrate-modified filter paper was not measurable. | ||
| Scheme 2 A synthetic pathway for the preparation of QPDMAEMA-grafted filter paper. | ||
The 2-bromoisobutyrate-functionalized filter paper was then subjected to SIP of the DMAEMA monomer via a recently developed living radical polymerization process, activators regenerated by electron transfer for atom transfer radical polymerization (ARGET ATRP).46–48 This mode of polymerization has been chosen as a method for fabricating the positively charged membrane in this particular case due to the fact that the polymerization is living so the molecular weight or graft density of the attached polymer chains can be well controlled. More importantly, the reaction can be performed in a limited amount of air using a ppm level of a catalyst in the presence of a reducing agent without a need for rigorous deoxygenation. This simple handling strategy really opens up an opportunity to produce this paper-based membrane at a larger scale for future DNA sensor applications.
In this research, the ARGET ATRP of DMAEMA was conducted using the CuBr2–Me6TREN complex and Sn(EH)2 as a catalytic system and reducing agent, respectively. To monitor the molecular weight of the PDMAEMA formed in solution, the sacrificial initiator, EBiB, was added. For the target degree of polymerization (DP) at 200 ([DMAEMA]0/[EBiB]0 = 200:1), it was found that the [Sn(EH)2]0/[CuBr2]0 of 400 was the optimal ratio that gave the polymer with well-controlled characteristic (Mn (exp) = 28495 being close to a Mn (theo) of 28260 and PDI = 1.28) (Table S3, ESI† entry 3). Increasing the ratio to 667 did not provide a positive impact on the polymerization process (Table S3, ESI† entry 4). On the other hand, the PDI tended to be high at a ratio of 200. It should be emphasized that acetone is a better solvent for polymerization of DMAEMA using this catalytic system than anisole (Table S3, ESI† entry 1), the solvent used for the synthesis of PDMAEMA via ARGET ATRP using the CuCl2–tris[(2-pyridyl)methyl]amine (TPMA) complex as the catalyst, previously reported by others.46 This is quite desirable given that acetone is a non-toxic organic solvent having a low boiling point so it can be easily removed from the filter paper after the SIP process by air drying without the need for heat treatment.
The ability to control the molecular weight of PDMAEMA is demonstrated in Table 2. Apparently, the molecular weight of PDMAEMA correspondingly increases as a function of [DMAEMA]0/[EBiB]0 from 100:1 to 200:1 and 400:1 or a target DP of 100, 200, and 400, respectively. The PDI values approaching 1.0 suggest that the polymerization can be well controlled and gave PDMAEMA with narrow molecular weight distribution. GPC traces of the synthesized PDMAEMA having different target DP are displayed in Fig. S2, ESI.†
| Entry | Target DPa | % Convb | M n (theo)c | M n (exp)d | PDId |
|---|---|---|---|---|---|
|
a EBiB/CuBr2/Me6TREN = 1:0.006:0.25. The polymerization was conducted in a scintillation vial with volume of free space = 7.7 mL.
b Determined by 1H NMR in CDCl3.
c
M
n (theo) = ([DMAEMA]0/[EBiB]0) × conversion.
d Determined by GPC in THF, based on polystyrene standards.
|
|||||
| 1 | 100 | 85 | 13345 |
21010 |
1.24 |
| 2 | 200 | 90 | 28260 |
28495 |
1.28 |
| 3 | 400 | 69 | 43332 |
42650 |
1.18 |
Quaternization of PDMAEMA-grafted filter paper was accomplished by methylation of the amino groups of PDMAEMA on filter paper with methyl iodide at ambient temperature as shown in Scheme 2. The iodide counter ion must be exchanged for chloride by treatment with 0.1 M NaCl. This step is mandatory because it was found that iodide ion can be oxidized into iodine which makes the paper turn slightly yellowish upon storage. After it was air-dried, the QPDMAEMA-grafted filter paper having chloride as the counter ion was obtained. The filter paper became extremely hydrophilic after being grafted with PDMAEMA. Although a contact angle of ∼50° can be roughly estimated from the drop image promptly captured at the initial contact using the Adobe Photoshop CS2, the water drop was rapidly absorbed into the PDMAEMA-grafted filter paper and disappeared within a second after the contact. This is much more hydrophilic than that has been observed for PDMAEMA-grafted on relatively flat surfaces (∼50°).49,50 This wetting behavior remained unaltered after quaternization. Images of water droplets during water contact angle measurements of both samples are shown in Fig. S1b and c (ESI†).
As examined by SEM (Fig. S3, ESI†), there was no visible changes of surface morphology of the filter paper particularly after being grafted with PDMAEMA having a target DP of 200 (both before and after quaternization) implying that the layer of the coated polymer is relatively thin. This observation coincides with the fact that the physical strength and resistance to bending of the filter paper were not affected by polymer grafting. On the other hand, the filter paper became tougher and plastic-like upon grafting with PDMAEMA having a higher target DP of 400 (SEM micrograph not shown) suggesting that a thicker polymer layer was deposited on the filter paper.
The functional group identity of each surface-modified filter paper was determined by FT-IR as shown in Fig. S4 (ESI†). The FTIR spectra of the initiator immobilized filter paper (Fig. S4b, ESI†) show a band at ∼1728 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of 2-bromoisobutyrate ester) which is not present in the virgin filter paper (Fig. S4a, ESI†). After being grafted with PDMAEMA, the absorption peak for the carbonyl group appears at the same position with greater intensity (Fig. S4c, ESI†), which is a characteristic of the CO stretching of PDMAEMA.40,41 No significant change in the IR bands was observed after quaternization (Fig. S2d, ESI†).
O stretching of 2-bromoisobutyrate ester) which is not present in the virgin filter paper (Fig. S4a, ESI†). After being grafted with PDMAEMA, the absorption peak for the carbonyl group appears at the same position with greater intensity (Fig. S4c, ESI†), which is a characteristic of the CO stretching of PDMAEMA.40,41 No significant change in the IR bands was observed after quaternization (Fig. S2d, ESI†).
The elemental compositions of all surface-modified filter papers were evaluated by XPS and the corresponding spectra are shown in Fig. S5–S9 (ESI†). From the XPS atomic composition data illustrated in Table S4 (ESI†), the detection of 0.8% bromine on the initiator immobilized filter paper suggests that there are bromoester groups available for initiating polymerization from the surface of filter paper. A trace amount of bromine (∼0.2%) detected on the PDMAEMA- and QPDMAEMA-grafted filter papers indicated that there were still living polymer chain ends in the form of dormant bromoester species. The presence of an atomic signal of nitrogen on the PDMAEMA- and QPDMAEMA-grafted filter papers clearly confirms the successful polymer attachment via the SIP process. The C/N ratio of 8.1 (Table S4, ESI†) corresponds quite well with the theoretical value of PDMAEMA (8.0). An increase in C/N ratio to 11.9 together with the emergence of the N1s peak at 402.0 eV on the QPDMAEMA-grafted filter paper (Fig. 2) strongly indicates that additional carbon atoms have been introduced upon quaternization and that most nitrogen atoms existed in the less electronegative form of quaternary ammonium groups.42 The extent of surface quaternization estimated from the relative ratio between the area of the N1s from the quaternary ammonium entities and that of the regular, non-quaternized one appearing at 398.6 eV was about 80% (Fig. 2). This figure is relatively high suggesting that the QPDMAEMA-grafted filter paper should be positively charged irrespective of pH and readily available to electrostatically adsorb DNA. To test such adsorption ability, a synthetic 13 mer DNA corresponding to a partial sequence of human IL-10 promoter gene (DNA SLE2) was used as a model. A 1 μL of 10 μM DNA in 0.4 M NaOH containing 10 mM EDTA was spotted on the QPDMAEMA-grafted filter paper. XPS analysis of the paper after extensive washing to remove the unbound DNA (Table S4, ESI†) revealed a characteristic signal of the phosphate group, with a phosphorus content of 1.2%, implying that the DNA molecules were successfully adsorbed on the QPDMAEMA-grafted filter paper.
 | ||
| Fig. 2 N1s XPS spectra of PDMAEMA- (right) and QPDMAEMA- (left) grafted filter papers. | ||
DNA detection following Dot blot hybridization
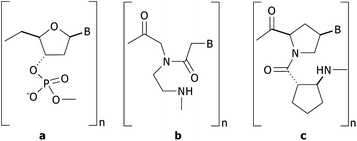
It should be emphasized that most of the tests were performed on the QPDMAEMA-grafted filter paper prepared using a target DP of 200. The results are displayed in two formats: scanned images (column A) and profile images as analyzed by Scion Image (column B). The profile image should provide the semi-quantitative intensity of the colorimetric readout and therefore reflecting the testing efficiency and facilitating the background and signal differentiation. A preliminary optimization was first carried out with 1 pmol of b-DNA SLE2 as a positive control by varying the enzyme, substrate, concentration and reaction time. The results shown in Fig. S10 (ESI†) suggest that SA–HRP was superior to avidin–HRP, and the optimal amount was 20 ng per spot since this condition provided a high signal and a minimal background due to non-specific adsorptions (Fig. S10d, ESI†). According to Fig. S11 (ESI†), the optimum concentration of the substrate mixture is 250 μL of 1.6 mg mL−1 OPD and 250 μL of 1.6 mg mL−1 urea–H2O2 and the optimum activation time is 1 min because this condition gave a high signal to background ratio (Fig. S11c, ESI†). Increasing the concentration of substrate mixtures and/or reaction time gave a stronger signal, but so as the non-specific adsorption (background). When TMB34,35 was used as a substrate in place of OPD, a stronger color was obtained, but also with higher background (Fig. S11a, ESI†). In addition, the blue color disappeared after storage at room temperature suggesting that the complex formed was not stable and that TMB was not the appropriate substrate. The use of a blocking solution (especially 1% BSA) has been found to be essential in preventing the non-specific interaction between the SA–HRP and QPDMAEMA-grafted filter paper as demonstrated in Fig. S12a (ESI†). As seen in Fig. S12b, ESI† signals from both expected specific hybridization and unwanted non-specific adsorption were largely suppressed after blocking with 1% skim milk, a common blocking reagent for various bioassays, suggesting that it is not a suitable blocking reagent.To test the ability of QPDMAEMA-grafted filter paper as a membrane for DNA sequence determination by Dot blot hybridization, two closely related synthetic 13 mer oligodeoxynucleotides (DNA SLE1 and DNA SLE2) differing by just one base were spotted onto the membrane and the sequences were detected by the b-PNA SLE2 probe. Details of DNA and PNA sequences and the description of samples/probes applied in each spot (1–6) are displayed in Tables S1 and S2 (ESI†), respectively. A biotinylated DNA (b-DNA SLE2) was included as a positive control to show the maximum signal that can be generated by the enzymatic reaction. The effects of washing and blocking solutions were examined using the optimal conditions mentioned above (see results in Fig. S10d, S11c, and S12a, ESI†). Only the positions with b-DNA SLE2 [(+)1)] and DNA SLE2/b-PNA SLE2 [(+)4] (Table S1, ESI†) show yellow spots visible to the naked eyes (Fig. 3). Lowering the PBS concentration down from 0.1 M to 50 mM in the absence of 0.1 M NaCl (Fig. 3d) gave inferior detection efficiency, with slight yellowish tints observed on the two negative controls [(−)5) and (−)6)]. Such non-specific adsorption disappeared and a good detection result was obtained (comparable to the result shown in Fig. 3b) upon addition of 0.1 M NaCl (Fig. 3c). For this reason, 0.1 M PBS without salt addition (Fig. 3b) was chosen as washing and blocking buffers for all subsequent experiments.
 | ||
| Fig. 3 Representative images (column A: scanned image and column B: profile image) of the test results demonstrating the effect of PBS concentration and salt addition in washing and blocking solutions: (a) 0.1 M PBS w/ 0.1 M NaCl, (b) 0.1 M PBS w/o 0.1 M NaCl, (c) 50 mM PBS w/ 0.1 M NaCl, and (d) 50 mM PBS w/o 0.1 M NaCl. | ||
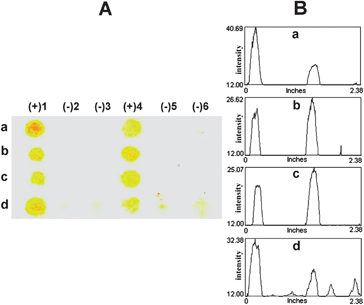
Three acpcPNA probes, b-PNA SLE2 and b-PNA SLE1, which differ by only one base, and an unrelated b-PNA (TG) were chosen for specificity determination. Details of DNA and PNA sequences and the description of samples/probes applied in each spot (1–6) are illustrated in Tables S1 and S5 (ESI†), respectively. The data presented in Fig. 4a and b reveal that the Dot blot hybridization using the QPDMAEMA-grafted filter paper and acpcPNA probes offers an excellent specificity to distinguish single base mismatches in the DNA target consisting a partial sequence of human IL-10 promoter gene (SLE1 and SLE2) by showing only the signal of b-PNA that was hybridized with the complementary DNA target. In addition to these two sequences, the same technique employing b-PNA (TG) as a probe can also successfully distinguish between the complementary and non-complementary DNA targets since the yellow spot was only observed at the position of the complementary DNA target (AC) (Fig. 4c). This illustrates the general applicability of the membrane for detection of various DNA sequences.
 | ||
| Fig. 4 Representative images (column A: scanned image and column B: profile image) of the test results demonstrating the specificity and efficiency of (a) b-PNA SLE2, (b) b-PNA SLE1 to distinguish complementary and single mismatched DNA (SLE2 and SLE1) targets or (c) complementary DNA (AC) and non-complementary DNA (AG) using b-PNA (TG). The positive results are shown by the yellow spots at position 4 for the tests in entries a and b, and 2 for entry b. | ||
For the results shown in Fig. 5–7, details of DNA and PNA sequences and the description of samples/probes applied in each spot (1–6) are displayed in Tables S1 and S2 (ESI†), respectively. To compare the performance of the modified filter paper described in this work with commercial membranes, the detection of DNA SLE2 with b-PNA SLE2 was selected for the investigation. The results presented in Fig. 5 showed that only the QPDMAEMA-grafted filter paper (Fig. 5d) provided a clear-cut discrimination between the complementary [(+)4] and single mismatched DNA [(−)2]. On the other hand, the unquaternized PDMAEMA-grafted filter paper (Fig. 5c) gave low signals even with the positive control [(+)1], indicating that the quaternary ammonium groups of QPDMAEMA are essential for the efficient capture of DNA. The commercial nitrocellulose (Fig. 5a) and Nylon 66 (Fig. 5b) membranes showed substantial non-specific interactions as shown by the presence of dark spots in places where negative results should be observed. In the case of nitrocellulose (Fig. 5a), the appearance of signals whenever the b-PNA was applied, even in the absence of the correct DNA target [(−)2 and (−)5], suggested that there were non-specific interactions between the surface of the membrane and the b-PNA probe. With Nylon 66 (Fig. 5b), the colored spots were observed in all six positions implying that there were non-specific interactions between the membrane surface and SA–HRP as well.
 | ||
| Fig. 5 Representative images (column A: scanned image and column B: profile image) of the test results demonstrating the performance of the QPDMAEMA-grafted filter paper in comparison with commercial membranes: (a) nitrocellulose, (b) Nylon 66, (c) PDMAEMA-, and (d) QPDMAEMA-grafted filter paper. | ||
To test the stability of the QPDMAEMA-functionalized filter paper upon storage, the Dot blot experiments were carried out using a membrane that has been stored at room temperature (27–33 °C) for 1 month in comparison with the freshly prepared membranes. The results shown in Fig. 6 demonstrate that both membranes provided comparable results in distinguishing complementary and mismatched DNA targets suggesting that the modified filter paper is stable and the detection efficiency is not affected upon storage for at least one month. This outcome is quite desirable from the practical perspective.
 | ||
| Fig. 6 Representative images (column A: scanned image and column B: profile image) of the test results demonstrating sequence determination of the DNA SLE2 sample using the b-PNA SLE2 probe on the QPDMAEMA-grafted filter paper: (a) freshly prepared and (b) stored at room temperature (27–33 °C) for 1 month. | ||
The lowest amount of DNA that can still allow a clear discrimination between fully complementary (DNA SLE2) and single-mismatched DNA (DNA SLE1) was evaluated by spotting different quantities of the DNA targets (2 pmol to 10 fmol) followed by the detection using 2 pmol of the b-PNA SLE2 probe. The results in Fig. 7 show that unambiguous discrimination, which can be quoted as the detection limit of this colorimetric method, is at least down to 10 fmol (equivalent to 1 μL of 10 nM DNA). Although this detection limit is somewhat higher than some other reports,32,34,35 our main objective to develop a positively charged filter paper-based membrane for simple naked eye detection of DNA using a PNA probe and rapid analysis of multiple DNA samples with a short reaction time (less than 1 hour from DNA spotting to color readout) has been fulfilled. We have previously demonstrated that this level of sensitivity is more than sufficient to detect DNA samples from standard PCR products24 and, if necessary, it should be possible to improve the detection limit further using polybiotin tags35,51,52 or polymeric enzymes.35
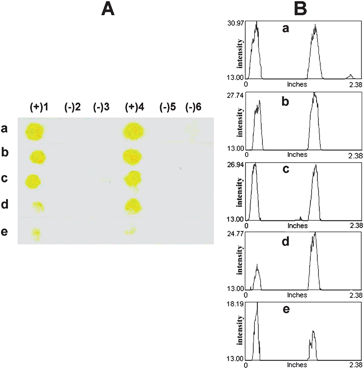 | ||
| Fig. 7 Representative images (column A: scanned image and column B: profile image) of the test results demonstrating the detection limit of complementary DNA SLE2 and single mismatched DNA SLE1 using 2 μL of 1 μM b-PNA SLE2 as the probe: (a) 2 pmol (2 μL of 1 μM), (b) 1 pmol (1 μL of 1 μM), (c) 100 fmol (1 μL of 100 nM), (d) 50 fmol (0.5 μL of 100 nM), and (e) 10 fmol (1 μL of 10 nM). | ||
The limitation of this present technique is similar to all blotting techniques that use non-specific interaction to immobilize DNA. If the DNA target to be detected is contaminated by other DNA sequences, there will be a competition between different DNA targets in binding to the membrane and the desired sequence may not be detectable. In such a scenario, a sandwich assay in which two acpcPNA probes that bind to different regions of the DNA target – one probe is immobilized on the surface for capturing the DNA target and the other is labeled with enzyme for detection – should solve the problem.35
Conclusions
QPDMAEMA was successfully grafted onto the filter paper by surface-initiated polymerization of DMAEMA via ARGET ATRP followed by quaternization with methyl iodide. The molecular weight and polydispersity index of the polymer can be controlled by adjusting the ratio of the Sn(EH)2/initiator and reaction time. The QPDMAEMA-grafted filter paper showed better performance than Nylon 66 and nitrocellulose to be used as a membrane for DNA sequence detection following a Dot blot format employing a biotinylated acpcPNA probe and an enzyme-mediated colorimetric assay. The low non-specific interaction of the modified filter paper, together with the high specificity of the PNA probe, allows single mismatch discrimination down to 10 fmol of DNA targets under non-stringent conditions.Acknowledgements
The financial support for this project was provided by Research Team Consolidation Grant, the Thailand Research Fund (RTA 5280002), the National Research University Project of Commission of Higher Education (CHE) and the Ratchadapiseksomphot Endowment Fund (Project Code AM1006A), and the Thai Government Stimulus Package 2 (TKK2555), under the Project for Establishment of Comprehensive Center for Innovative Food, Health Products and Agriculture. The authors are indebted to Dr Jack Hirsch of Polymer Science and Engineering Department, University of Massachusetts, Amherst, for assistance with XPS analysis. PSL acknowledges Kasetsart University, Chalermphrakiat Sakon Nakhon Province Campus and the Development and Promotion of Science and Technology Talents Project (DPST) for a PhD scholarship.References
- C.-P. Chak, J. M. Y. Lai, K. W. Y. Sham, C. H. K. Cheng and K. C.-F. Leung, RSC Adv., 2011, 1, 1342 RSC.
- J. Baur, C. Gondran, M. Holzinger, E. Defrancq, H. Perrot and S. Cosnier, Anal. Chem., 2009, 82, 1066 CrossRef.
- A. Akane, S. Seki, H. Shiono, H. Nakamura, M. Hasegawa, M. Kagawa, K. Matsubara, Y. Nakahori, S. Nagafuchi and Y. Nakagome, Forensic Sci. Int., 1992, 52, 143 CrossRef CAS.
- H. Pfitzinger, B. Ludes and P. Mangin, Int. J. Leg. Med., 1993, 105, 213 CrossRef CAS.
- M. Passamano and M. Pighini, Sens. Actuators, B, 2006, 118, 177 CrossRef.
- B. Kannan, D. E. Williams, M. A. Booth and J. Travas-Sejdic, Anal. Chem., 2011, 83, 3415 CrossRef CAS.
- H. Yin, Y. Zhou, C. Chen, L. Zhu and S. Ai, Analyst, 2012, 137, 1389–1395 RSC.
- K.-H. Lee, J. O. Lee, S. Choi, J.-B. Yoon and G.-H. Cho, Biosens. Bioelectron., 2012, 31, 343 CrossRef CAS.
- S. Hason, H. Pivonkova, V. Vetterl and M. Fojta, Anal. Chem., 2008, 80, 2391 CrossRef CAS.
- X. Tang, H. Liu, B. Zou, D. Tian and H. Huang, Analyst, 2012, 137, 309 RSC.
- Z. Gao, Analyst, 2012, 137, 1674 RSC.
- D. Berdat, A. Marin, F. Herrera and M. A. M. Gijs, Sens. Actuators, B, 2006, 118, 53 CrossRef.
- M. Kaatz, H. Schulze, I. Ciani, F. Lisdat, A. R. Mount and T. T. Bachmann, Analyst, 2012, 137, 59 RSC.
- M. Liu, X. Lou, J. Du, M. Guan, J. Wang, X. Ding and J. Zhao, Analyst, 2012, 137, 70 RSC.
- X. Mao, L. Yang, X.-L. Su and Y. Li, Biosens. Bioelectron., 2006, 21, 1178 CrossRef CAS.
- M. Egholm, O. Buchardt, L. Christensen, C. Behrens, S. M. Freier, D. A. Driver, R. H. Berg, S. K. Kim, B. Norden and P. E. Nielsen, Nature, 1993, 365, 566 CrossRef CAS.
- P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Science, 1991, 254, 1497 CAS.
- C. Suparpprom, C. Srisuwannaket, P. Sangvanich and T. Vilaivan, Tetrahedron Lett., 2005, 46, 2833 CrossRef CAS.
- T. Vilaivan and C. Srisuwannaket, Org. Lett., 2006, 8, 1897 CrossRef CAS.
- C. Ananthanawat, T. Vilaivan and V. P. Hoven, Sens. Actuators, B, 2009, 137, 215 CrossRef.
- C. Ananthanawat, V. P. Hoven, T. Vilaivan and X. Su, Biosens. Bioelectron., 2011, 26, 1918 CrossRef CAS.
- C. Ananthanawat, T. Vilaivan, V. P. Hoven and X. D. Su, Biosens. Bioelectron., 2010, 25, 1064 CrossRef CAS.
- C. Ananthanawat, T. Vilaivan, W. Mekboonsonglarp and V. P. Hoven, Biosens. Bioelectron., 2009, 24, 3544 CrossRef CAS.
- B. Boontha, J. Nakkuntod, N. Hirankarn, P. Chaumpluk and T. Vilaivan, Anal. Chem., 2008, 80, 8178 CrossRef CAS.
- M. A. Cook, A. M. Osborn, J. Bettandorff and P. A. Sobecky, Microbiology, 2001, 147, 2089 CAS.
- M. S. Urdea, B. D. Warner, J. A. Running, M. Stempien, J. Clyne and T. Horn, Nucleic Acids Res., 1988, 16, 4937 CrossRef CAS.
- E. Heinicke, U. Kumar and D. G. Munoz, J. Immunol. Methods, 1992, 152, 227 CrossRef CAS.
- M. Musiani, M. Zerbini, D. Gibellini, G. Gentilomi, S. Venturoli, G. Gallinella, E. Ferri and S. Girotti, J. Clin. Microbiol., 1991, 29, 2047 CAS.
- M. Schäferling and S. Nagl, Anal. Bioanal. Chem., 2006, 385, 500 CrossRef.
- N. Graf, S. Kassube and R. Krämer, Bioorg. Med. Chem. Lett., 2008, 18, 4786 CrossRef CAS.
- N. Graf and R. Krämer, Chem. Commun., 2006, 4375 RSC.
- J. J. Leary, D. J. Brigati and D. C. Ward, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 4045 CrossRef CAS.
- M. Renz and C. Kurz, Nucleic Acids Res., 1984, 12, 3435 CrossRef CAS.
- X. Su, H. F. Teh, X. H. Lieu and Z. Q. Gao, Anal. Chem., 2007, 79, 7192 CrossRef CAS.
- N. Zhang and D. H. Appella, J. Am. Chem. Soc., 2007, 129, 8424 CrossRef CAS.
- C. Kemper, K. Berggren, Z. Diwu and W. F. Patton, Electrophoresis, 2001, 22, 881 CrossRef CAS.
- K. C. Reed and D. A. Mann, Nucleic Acids Res., 1985, 13, 7207 CrossRef CAS.
- C.-M. Cheng, A. W. Martinez, J. Gong, C. R. Mace, S. T. Phillips, E. Carrilho, K. A. Mirica and G. M. Whitesides, Angew. Chem., Int. Ed., 2010, 49, 4771 CrossRef CAS.
- A. W. Martinez, S. T. Phillips, G. M. Whitesides and E. Carrilho, Anal. Chem., 2010, 82, 3 CrossRef CAS.
- S. B. Lee, R. R. Koepsel, S. W. Morley, K. Matyjaszewski, Y. J. Sun and A. J. Russell, Biomacromolecules, 2004, 5, 877 CrossRef CAS.
- D. Roy, J. S. Knapp, J. T. Guthrie and S. Perrier, Biomacromolecules, 2008, 9, 91 CrossRef CAS.
- F. Zhou, Z. Zheng, B. Yu, W. Liu and W. T. S. Huck, J. Am. Chem. Soc., 2006, 128, 16253 CrossRef CAS.
- M. Ciampolini and N. Nardi, Inorg. Chem., 1966, 5, 41 CrossRef CAS.
- P. Akkahat and V. P. Hoven, Colloids Surf., B, 2011, 86, 198 CrossRef CAS.
- A. Carlmark and E. Malmstrom, J. Am. Chem. Soc., 2002, 124, 900 CrossRef CAS.
- H. Dong and K. Matyjaszewski, Macromolecules, 2008, 41, 6868 CrossRef CAS.
- K. Matyjaszewski, H. C. Dong, W. Jakubowski, J. Pietrasik and A. Kusumo, Langmuir, 2007, 23, 4528 CrossRef CAS.
- K. Matyjaszewski, W. Jakubowski, K. Min, W. Tang, J. Y. Huang, W. A. Braunecker and N. V. Tsarevsky, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15309 CrossRef CAS.
- S. Gupta, M. Agrawal, M. Conrad, N. A. Hutter, P. Olk, F. Simon, L. M. Eng, M. Stamm and R. Jordan, Adv. Funct. Mater., 2010, 20, 1756 CrossRef CAS.
- J.-K. Chen and B.-J. Bai, J. Phys. Chem. C, 2011, 115, 21341 CAS.
- A. Emileh, E. Vasheghani-Farahani and M. Imani, Eur. Polym. J., 2007, 43, 1986 CrossRef CAS.
- A. H. Al-Hakim and R. Hull, Biochem. J., 1988, 251, 935 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: PNA and DNA sequences, GPC traces, water contact angles, SEM images, FTIR spectra, XPS spectra and atomic composition of the surface-modified filter papers, and additional test results. See DOI: 10.1039/c2an36133g |
| This journal is © The Royal Society of Chemistry 2013 |