Electrogenerated chemiluminescence of nanomaterials for bioanalysis
Shengyuan Deng and Huangxian Ju*
State Key Laboratory of Analytical Chemistry for Life Science, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, P.R. China. E-mail: hxju@nju.edu.cn
First published on 26th September 2012
Abstract
Since the electrogenerated chemiluminescence (ECL) of silicon nanoparticles (NPs) was reported in 2002, miscellaneous nanomaterials with various sizes and shapes have been employed as ECL nanoemitters for bioanalysis. Elucidation of the ECL derivation from these nanoemitters and pertinent biofunctionalization with multitudinous biomolecules can offer excellent ECL signal-transduction platforms for fabricating novel biosensing devices. In this review, we comprehensively describe retrospective and recent advances in NPs-based ECL and related biosensing methodologies, and review their analytical applications in the detection of small biological molecules, enzymatic sensing, immunoassay, DNA analysis and cytosensing.
 Shengyuan Deng | Shengyuan Deng is currently a PhD student in the School of Chemistry and Chemical Engineering, Nanjing University and has been working in Professor Ju's research group for 5 years. He received his BS in Chemistry from Kuang Yaming Honors School of Nanjing University in 2007. His current research involves synthesis and ECL behaviors of quantum dots, as well as their bioanalytical applications. |
 Huangxian Ju | Huangxian Ju received his BS, MS and PhD degrees from Nanjing University during 1982–1992. He was a post-doc. in Montreal University (Canada) from 1996 to 1997 and a guest professor in three universities of Germany and Ireland in 1999–2000. He became an associate and full professor of Nanjing University in 1993 and 1999. He won the National Funds for National Distinguished Young Scholars in 2003 and National Creative Research Groups in 2005, and was selected as a Changjiang Professor by the Education Ministry of China in 2007. His research interests focus on analytical biochemistry and molecular diagnosis. He has published 408 papers with an h-index value of 53, authored 24 patents, 2 English books, 6 Chinese books and 6 Chinese and 8 English chapters. |
1. Introduction
Electrogenerated chemiluminescence (electrochemiluminescence, ECL) is an electrochemically triggered optical radiation process produced by the energy relaxation of excited species.1 This technique intrinsically represents a marriage between electrochemistry and spectroscopy. By integrating both advantages, ECL possesses unique superiorities over other optical methods. First and foremost, it does not require a light source, which not only simplifies the detection apparatus but also invalidates background signals from scattered light and luminescent impurities, thus leading to high sensitivity. Secondly, ECL has a high specificity due to the emitter–coreactant relationship and good selectivity as the excited states can be regulated by alternating the applied potential. Therefore, the development of ECL-emitting species initiated with Grignard reagents and luminol2,3 have attracted continuous interest. Different polyaromatic hydrocarbons (PAHs)4 and organometallic compounds5,6 have been used as general ECL emitters due to their ECL natures, and the ECL technique has also become a powerful analytical tool in the detection of biomolecules, clinical diagnostics, environmental and food monitoring, and biowarfare agent detection, etc.7,8Recently, semiconductor nanocrystals or quantum dots (QDs) have been exploited as a new kind of ECL emitters since the ECL study of silicon nanoparticles (NPs) was first reported in 2002.9 Compared with conventional molecular emitters, QDs own several distinctive merits like size/surface-trap controlled luminescence and good stability against photobleaching. QDs-based ECL has accordingly been widely applied in biosensing and bioanalysis.10–12 As finding new luminophores with a high ECL efficiency for bioanalysis is the constant driving force of this area, the family of nanoemitters for ECL has been enlarged from exclusively QDs to other miscellaneous nanomaterials in recent years, with various compositions, sizes and shapes, including metallic nanoclusters,13–15 carbon nanodots,16–18 metallic oxide semiconductors,19–22 and even organic nano-aggregates.23–26 Further elucidation of the ECL derivation from these nanoemitters also promotes the development of novel ECL signal-transduction platforms for biosensing devices.
The advances concerning NPs-based ECL not only open a promising field for the development of new-generation ECL-emitting species, but also complement the conventional optical utilizations of QDs. This review covers the principles and related biosensing methodologies of NPs-based ECL, and examines their analytical applications the in detection of small biological molecules, enzymatic sensing, immunoassay, DNA analysis and cytosensing.
2. Kinetics of NPs-based ECL emission
Before the first report on ECL from nanocrystals in an organic solvent,9 the ECL phenomena of PAHs4 and organometallic complexes5,6 had been elaborated. Considering the similarity in frontier orbital theory with band theory of solids in energy level and electrochemical reactivity, the kinetics of NPs-based ECL can be related to two conventional courses: annihilation and coreactant courses.27–302.1. Annihilation course
The first NPs-based ECL emission of SiNPs was realized through annihilation.12 It is technically generated by redox cycling9,27–30 or differential pulsing.14,31,32 The conduction band and the valence band of an individual NP can accept (electron-injected, R˙−) and donate (hole-injected, R˙+) electrons under electrochemical conditions, respectively, to produce a couple of the single NP anion and cation radicals. The ECL emission then occurs following a comproportionation reaction by radical collision and recombination into the excited state (R*).8 The ECL emission of NPs such as CdSe,27 CdTe,29 PbS QDs,33etc., homogeneously dispersed in a deoxygenated organic phase normally follows this course with qualitative reaction equations:| R − e → R˙+ | (1) |
| R + e → R˙− | (2) |
| R˙− + R˙+ → R* + R | (3) |
| R* → R + hν | (4) |
Because the electron transfer from the electrode to the core of thiol-capped QDs charges the sub-attofarad capacitance of the thiol ligands – so-called quantized double-layer charging34 – no ECL is observed through annihilation-type kinetics for thioglycerol-capped CdS QDs. Therefore, another inconvenience of this type of ECL emission lays with its strong dependence on electrochemically inert moieties. Given that high over-potentials should be applied to overcome the surface energy of the NPs,9 this being time-consuming also restricts the application and sampling throughput. To date, only QDs with a superlattice structure have shown the necessary ECL applications.37
2.2. Coreactant course
The coreactant is a heterogeneously introduced species that, upon preceding oxidation or reduction, react with the electrolytic ECL luminophores to produce R*.7,8 Distinct from the annihilation route which demands both R˙+ and R˙−, the coreactant route experiences electron transfer between either R˙+ or R˙− and the coreactant. Generally, the coreactant route is an electrochemical (E) procedure followed by a chemical (C) one, which can, overall, be taken as an EC process.Bard and co-workers have firstly demonstrated that the SiNPs solution could achieve a higher ECL intensity by adding an excess of C2O42− into the solution.9 It was observed that ECL was initiated with the injection of holes into the SiNPs; meanwhile, the oxidation of C2O42− produced a strong reductant CO2˙−, which injected electrons into the LUMO of the oxidized SiNPs to produce R* then emitting ECL irradiation. Recent work has further proved that C2O42− can also work as an effective ECL coreactant for 9,10-diphenylanthracene nanorods.38
As the operation of the coreactant-type ECL emission features a unidirectional potential scan, this coreactant-participating kinetics can be further be divided into anodic and cathodic ECL emission, in which the coreactant correspondingly acts as the reductant and the oxidant, respectively. Thus the EC kinetics can further be simply designated as oxidative-reduction for anodic ECL emission and reductive-oxidation for cathodic ECL emission.7,8
So far, several substrates have been validated experimentally as potent coreactants in NPs-based ECL emission, among which some have already accomplished practical applications. Based on the bond cleavage position of the molecular structure during the ECL reaction, classic coreactants can be apparently divided into three groups: (a) organic amines, such as tri-n-propylamine (TPrA),39,40 dibutylaminoethanol (DBAE),40,41 triethanolamine (TEA),15,40,42etc.; (b) homolytic peroxides, such as O2,43,44 H2O2,45–47 S2O82−,48 and C2O42−;49 and (c) other small molecules like SO32−,50 and CH2Cl2.29 Nevertheless, their intermediates are the real participants for generating the excited states of NPs just as the superoxide radical for O2,44 and the hydroxyl radical for H2O2.47 In this way, each coreactant is appropriate to either anodic or cathodic ECL emission according to the valency and the electrode potential of the corresponding intermediate.
The coreactant course possesses several advantages over the annihilation path. For example, the coreactant is especially efficient when either R˙+ or R˙− is not quite stable enough for ECL of the NPs reaction or when the electrode or solvent has a narrow potential window so that R˙+ or R˙− cannot be formed; the unipolar potential step techniques spare considerable time and promote the measuring throughput; and an intense and sensitive ECL signal can be obtained, etc. The coreactant route is of great importance for biosensing applications of NPs-based ECL. Modern ECL applications of QDs are almost exclusively based on coreactants.51,52 However, the introduction of coreactants, especially the coreactant-related physicochemical behaviors like diffusion,15,40,42 and absorption,15,33 besides its concentration, inevitably complicate the ECL systems and greatly affect the rate-determining step of the ECL kinetics, which appears as changes in the plot profile and peak potential, especially for TPrA42 and S2O82−.14,53 The direct ECL emission from the implicit coreactant should also be taken into consideration as an experimental blank or control.54,55 In other words, a number of criteria need to be met for a qualified coreactant of NPs-based ECL,7 including solubility, stability, electrochemical activity, kinetics, ECL background, etc. Moreover, the coreactant should be easily oxidized or reduced at or near the electrode and undergo a rapid chemical reaction to form an intermediate as a strong reductant or oxidant to react with the oxidized or reduced NPs to form R*.8
Recently, a classification of the coreactant course has been presented in view of the sizes (molecular or nano-sized) of luminophores (L) and coreactants (C). The NPs-based ECL coreactant course is divided into two types: (a) LNano − CMol and (b) LNano − CNano.56 Metallic oxide semiconductors (MOS) like SnO56 and TiO257 have been reported as good candidates as solid coreactants of type (b) by offering metastable states for ECL electron hopping.
3. Derivation of NPs-based ECL emission
In view of crystallography, an individual nanosized particle can be divided into two crystalline regions. One is the bulk phase (the core) where metallic and non-metallic ions align periodically in long-range order; the other is the surface domain with an asymmetric chemical environment resulting from unpaired electrons,13,15 dangling bonds,16,58 ligand coordination,59,60 and crystalline defects.61,62 According to Bloch theorem, different structures correspond to disparate electronic states as well as their level distribution. Hence, two basic models for illustrating the derivation of NPs-based ECL irradiation are proposed as the surface-state model28 and the band-gap model.293.1. Surface-state model
As an ECL course is intrinsically electro-driven, the electrons exchange between the electrode and NPs via their surface-states. Given the large surface-area-to-volume ratio of NPs, a considerable proportion of energy bands are ascribed to the surface atoms, which derives the surface-state ECL emission as illuminated in Fig. 1.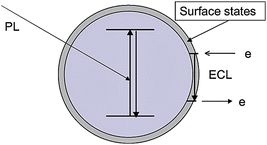 | ||
| Fig. 1 Schematic representation of PL and surface-state ECL emissions in a semiconductor NP. (Reproduced from ref. 28. Copyright 2003, American Chemical Society.) | ||
Despite the difference in the stimulating source, both ECL and photoluminescence (PL) kinetics culminate in light emission accompanied by the transition of electrons from the excited states to the ground states. Making comparisons between PL and ECL spectroscopies can attain a better understanding about the surface-state ECL. Due to the quantum size effect, PL mainly occurs through excitation and emission from the core, whereas ECL is susceptible to the surface states. Therefore, a surface-state ECL spectrum features an obvious red-shifted emission wavelength with respect to the PL spectrum.9,27 The reason is that wave functions concerning the states of electrons and holes at the surface superpose strongly, leading to a relatively narrower band-gap than that of the core.9 This red-shifted behavior has been observed at the ECL emission of SiNPs,9 GeNPs30 and CdSe QDs.27 Since the doping of heteroatoms and the introduction of coreactants both have impacts on the surface states, NPs-based ECL spectra sometimes display multiple peaks corresponding to these sophisticated interacting details,28,63,64 especially the ‘dual-peak’ which manifests that the surface states of the individual NPs are susceptible to coreactants.45,59,60 These phenomena are quite different from the ECL molecular spectrum.
The surface states also contribute to the PL spectrum. For example, an asymmetric PL emission peak with a non-zero background tail normally indicates the surface traps;27 a slow decay but strong correlation component in the fitted time-resolved PL curve represents the surface defects of NPs that trap electrons at the conduction band and generate a new excited state.65 These details in PL spectra reflect the relationship of PL and ECL, and demonstrate the possible surface-state ECL emission. In addition, the inconspicuous extinction absorption peak in the UV-vis spectrum can be attributed to the surface traps on the NPs.66 In other words, ECL and electrochemistry studies are sensitive to the surface states and have become a powerful tool for probing the surface chemistry and charge transfer dynamics,33 whereas PL spectroscopy specializes in exploiting the inner information of the band-gap transition.
Since the surface states generally show narrower band-gaps compared with the core, the surface-derived ECL emission possesses several advantages over the band-gap-derived type. The ECL signal always appears at a longer wavelength with a lower overpotential, which is beneficial for maintaining the bioactivity of the detection targets and excluding the interference from the coexisting electroactive species.66,67 The presence of surface traps makes the electron transfer between the NP surface and coreactant or electrode easier and further facilitates the disintegration of electron/hole-injected traps,68,69 resulting in a stronger ECL emission. These advantages lead to sensitive yet low-potential ECL emission.
Artificially manipulating the surface-states of NPs is the most direct idea for harvesting the surface-state ECL emission. Alternating the stoichiometric ratio of substrates is an empirical tryout, whereas intentionally selecting or arranging specific moieties has been confirmed as a practical implementation. For example, the rigid conformation from two cis-carboxyl groups of meso-2,3-dimercaptosuccinic acid (DMSA) can create a large amount of surface traps on CdTe QDs,66 and dual ligands such as mercaptopropionic acid (MPA) and sodium hexametaphosphate, and trioctylphosphine oxide (TOPO) and dodecylamine28,33 can hinder the formation of a well-coated QD shell, leading to a great number of surface traps. Surface-state ECL emissions have also been observed at uncalcinated MOS like TiO2-19,20,22,61 and CeO2-modified62 electrodes owing to excessive Frenkle or Shottky defects on their surfaces.
However, the desorption of ligands and leakage of uncapped heavy metallic ions due to the solvation effect have long been a problem for NPs with surface traps,70 which is especially prominent for water-soluble NPs with respect to their preparation methods. As the free ligands in the as-prepared solution have a side-effect on the optical property of NPs,70 these NPs show low quantum yields as well as optical and storage instability without thorough purification like ultrafiltration or dialysis prior to the ECL application.
3.2. Band-gap model
The band-gap model is another basic derivation type of NPs-based ECL and namely corresponds to the bulk of the NPs. The ECL spectrum matches the PL spectrum, and is size-dependent and tunable due to the dielectric confinement effect.Because electron-/hole-injection in NPs is generally assumed to occur via the surface states, the surface-state model has been considered as the main generation model for NPs-based ECL. Reports on ECL emission derived from the band-gap are relatively fewer.21,29,31,38,39 Bard and colleagues first presented that the surface states of CdTe QDs could be easily passivated to produce band-gap ECL (Fig. 2).28 Recently, the band-gap ECL emissions of thioglycolic acid-stabilized CdSe QDs and mercaptopropionic acid-capped CdTe QDs in aqueous solution have also been discovered by Ju et al.47,71
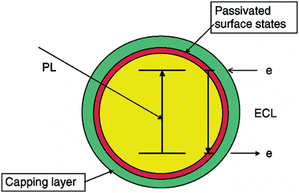 | ||
| Fig. 2 Schematic representation of PL and band-gap ECL emissions in a semiconductor NP. (Reproduced from ref. 28. Copyright 2003, American Chemical Society.) | ||
According to the structure rheology, the ECL derivation of certain NP species can vary from a surface-state type to a band-gap one or even remain both by unpassivating, totally or partially passivating the surface states, which has been validated through the ECL study of ZnSe@CdSe core–shell QDs.28 Unlike the ECL spectra of SiNPs or CdSe QDs with a single emission peak significantly red-shifted from the PL peak,9,27 ECL from ZnSe@CdSe QDs produces a spectrum containing two peaks: a sharp peak whose position is almost identical to that in the PL spectrum, and another broader peak with a red shift of ∼200 nm compared to that of the PL.28 This suggests emission from both surface states on the QDs and the bulk in QDs where the surface states have been passivated. The large ECL peak at the wavelength of band-edge PL suggests that the surfaces of ZnSe@CdSe QDs have been largely passivated, and ECL emission is mainly generated from the core of QDs.28 Nowadays, to overcome the above difficulties of surface-state NPs-based ECL emission, surface passivation has become a common consideration. Commercial QDs for PL emission are commonly synthesized in a core–shell structure like ZnS@CdSe QDs to passivate the surface traps largely for efficient PL intensity.51,72–75
The two generation types of NPs-based ECL have distinctive applications. The surface-state ECL is sensitive and can be used to probe the surface chemistry, while the band-gap ECL spectrum is size-dependent for multiplexed optical assays. Because the NPs-based ECL potential mainly depends on the surface-states and the conduction band edge, while being irrelevant to the band-gap,50,66,67,76–78 NPs with a small band-gap, especially near-infrared PL emission, do not assume low-potential ECL emission.33,79–81 Nonetheless, NPs-based ECL following the band-gap model has otherwise shown promising applications in immunoassay,82,83 DNA detection,84,85 and cytosensing.85
4. Coreactants for NPs-based ECL
The coreactants involved in NPs-based ECL mainly include four systems: peroxydisulfate system, hydrogen peroxide or oxygen system, tri-n-propylamine system and sulfite system.4.1. Peroxydisulfate system
Peroxydisulfate (S2O82−), long-since known as a coreactant of the Ru(bpy)32+–ECL system,48 can be used as a coreactant through so-called ‘reductive-oxidation’ for the ECL emission of SiNPs.9 The electrochemical reduction of S2O82− produces the intermediate SO4˙− with a strong oxidizing capacity, which can react with negatively-charged NPs by injecting a hole into the HOMO, inducing an excited state afterwards. The general mechanism for the S2O82− system is illustrated as follows:| R + e → R˙− | (5) |
| S2O82− + e → SO4˙− + SO42− | (6) |
| SO4˙− + R˙− → R* + SO42− | (7) |
4.2. Hydrogen peroxide system
Hydrogen peroxide (H2O2) is another important coreactant for NPs-based cathodic ECL. It was originally transplaced from the classic (electrogenerated) chemiluminescence system of luminol and H2O2.3,7,8 As the ubiquitous dissolved oxygen can be electrochemically reduced to H2O2, it also acts as an endogenous coreactant for ECL emission. Zou and Ju firstly demonstrated the electron-transfer reaction between electrochemically reduced CdSe QDs and H2O2 coreactant, which resulted in ECL emission from NPs.45 The general mechanism for the H2O2 (or O2) system is assumed as follows:| R + e → R˙− | (8) |
| O2 + H2O + 2e → HO2˙− + OH− | (9) |
| 2R˙− + HO2˙− + H2O → 3OH− + 2R* | (10) |
| 2R˙− + H2O2 → 2OH− + 2R* | (11) |
The dissolved oxygen in the detection solution is a ready-made coreactant. That NPs-based ECL solely relies on O2 without the introduction of exogenous coreactants makes the system neat, green and facile. A preliminary ECL test of unknown NPs just in the air-saturated solution can evaluate their ECL capability. Besides, as O2 and H2O2 both play key roles in the metabolism as well as in industrial reactions, it is of great interest to quantify them with the NPs-based ECL technique.19,20,22,45,47,102–107 As they are the substrates of oxidases and peroxidases, many NPs-based ECL biosensors have been fabricated for multifarious analytes by enzyme-catalytic cycles.43,66,67,86,98–100,108
4.3. Tri-n-propylamine system
Among organic amines, TPrA is a most popular coreactant for anodic ECL systems through ‘oxidative-reduction’.7,8 The major applications of TPrA reported so far involve Ru(bpy)32+ and its derivatives as ECL emitters and TPrA as the coreactant, which has become the commercial benchmark for ECL immunoassay and DNA analysis.8 ECL emission resulting from electro-oxidized rubrene aggregates and TPrA suggests that TPrA may also work as a coreactant for NPs-based ECL emission.26 Recently, anodic ECL emission of MPA-capped CdS@CdTe QDs with TPrA as the coreactant in aqueous solution has been demonstrated.77Electrochemical studies of various aliphatic amines indicate a possible reaction pathway for oxidizing TPrA to produce a strong reducing agent.39 Upon oxidation, the short-lived TPrA radical cation (TPrA˙+) is believed to lose a proton from an α-carbon to form the strongly reducing intermediate TPrA˙, and then reduces QD˙+ to QDs*. The general mechanism for the TPrA system is deduced as shown below:
| R − e → R˙+ | (12) |
| TPrA − e → TPrA˙+ | (13) |
| TPrA˙+ → TPrA˙ + H+ | (14) |
| R˙+ + TPrA˙ → R* + Pr2N+ | (15) |
4.4. Sulfite system
Sulfite (SO32−) can enhance the anodic ECL of MPA-capped CdTe QDs by the following mechanism:50| R − e → R˙+ | (16) |
| SO32− − e → SO3˙− | (17) |
| 2OH− + SO3˙− + 2O2 → O2˙− + SO42− + H2O | (18) |
| R + O2˙ → R˙− + O2 | (19) |
| R˙+ + R˙− → R* | (20) |
5. Miscellaneous ECL nanoemitters
NPs-based ECL systems can be classed by the different features of emitters, such as composition, volume and morphology. Here, we categorize various kinds of nanoemitters, mainly by their compositions.5.1. Binary component quantum dots
The binary component QDs as ECL emitters mainly include CdS,34,111 CdSe27 and CdTe.29,112,113 These QDs are composed of metallic elements of the I and II subgroups and non-metallic elements of the IV and VI main groups. The ECL behaviors of other QDs such as ZnS,114 ZnSe,115 PbS,33,116 CuSe,117 CdO,118 and Bi2Te3,119,120 have been extensively studied. Among them, ZnS has drawn much attention for in vitro low toxicity.63,80,81,83–855.2. Core–shell structured quantum dots
As non-radiative dissipation derived from the surface traps can be inhibited at its maximum by surface passivation to produce a highly improved ECL performance, especially in the stability and quantum yield,28,81 core–shell or yolk–shell structured QDs such as ZnSe@CdSe,28 ZnS@CdSeTe79,80 and ZnS@CdSe84,85 have emerged by capping with environmentally-friendly elements to prevent leakage of toxic ions. The shell components can simultaneously adjust the wavelength and intensity of the ECL emission, which has been observed for CdS@CdTe,77 CdS@CdSe,82,83 and CdS@ZnO QDs.121 Based on this behavior, the primary singular shell is further extended to multilayered QDs, such as ZnS@CdS@CdTe QDs,81 which realizes a near-infrared (NIR) ECL emission.5.3. Doped quantum dots
Doping conventional QDs with other ions can generate unexpectedly interesting ECL behaviors. For example, incorporating Se into CdTe QDs successfully harvests the NIR ECL emission.79,80 The doping of a trace amount of Mn2+ produces a new yet minor ECL emission of ZnS QDs at a relatively low-potential (e.g. −1.5 V).63 Taking the merit of paramagnetic Mn2+, the Mn2+-doped CdS QDs possess magnetism, and the ECL emission shows magnetic resonance with Fe3O4 NPs though an opto-magnetic interaction.122 Interestingly, Mn(II) as the dopant inserts a transition state between the original valence and conduction bands of the QD, leading to an electro-phosphorescence emission by a triplet–triplet conversion upon excitation.122–129 The f-electrons of the rare-earth element Eu3+ induce a new emission peak around 620 nm in the ECL spectrum of the doped CdS QDs, which belongs to the 5D0→7F2 transition of the Eu3+ ions. The doping of Eu3+ ions causes a 4-fold enhancement in ECL intensity and shows a great O2 sensitivity benefiting from the high oxygen affinity of Eu3+.1305.4. Single element nanoparticles
Single element semiconductor NPs such as Si9 and Ge30 have taken an irreplaceable spot in the history of NPs-based ECL since the ECL phenomenon of SiNPs was marked as its origin. Recently this family of monocomponent ECL nanoemitters has been quickly developed due to the observation of ECL emission from Ag13 and Au14,15,131 nanoclusters (NCs). These NCs have a large proportion of surface atoms with different valence states like Au(III) and Au(V), which consist of special surface states (Fig. 3).132,133 The ECL of AuNCs has shown an anomalous response to dopamine14 and H2O2,131 resulting in ‘signal-on’ and ‘signal-off’ detection, respectively, which are the opposite of the tendencies measured at conventional QDs, implying quite different mechanisms.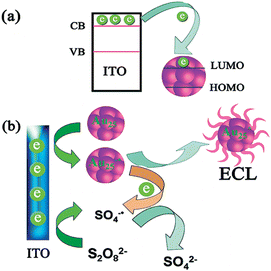 | ||
| Fig. 3 Schematic illustration of (a) electron transfer between ITO and Au NCs and (b) the ECL mechanisms of Au NCs. (Reproduced from ref. 14. Copyright 2011, American Chemical Society.) | ||
The discovery of ECL from carbon NCs or nanodots is also a breakthrough as they are is superior in water-solubility, being environmentally friendly, biocompatible and biofunctionalizable with various capping agents.13–15,131–133 Different carbon allotropes from micro- to macro-scope have been verified as the source of carbon NCs.16–18,134 Meanwhile, the ECL behavior of graphene oxide (GO) has also been investigated (Fig. 4).58 Although single element NPs are state-of-the-art ECL nanoemitters, a high applying potential and a low emission intensity are their defects, which limits the applications of these nanoemitters through a less acceptable sensitivity of measurement.14,131 As AgNCs can be in situ incorporated into the duplex of double-stranded DNA135–137 and possess a high cumulative stability constant of metal coordiantes,138 single element ECL nanoemitters are expected to have promising application in the development of detection methodology.
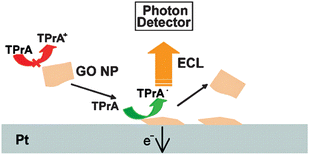 | ||
| Fig. 4 Schematic representation of the ECL emissions in GO NP. (Adapted from ref. 58. Copyright 2009, American Chemical Society.) | ||
5.5. Metallic oxide semiconductors
MOS NPs are intrinsically similar to conventional QDs except for a comparatively wide band-gap. The ECL of TiO2 NPs, especially the effect of polymorphism on the ECL performance of anatase and rutile TiO2, has been well studied.19–22,61,139,140 AgNPs-modified TiO2 NTs show a sensitive ECL signal for the detection of PAHs with benzo(a)pyrene as the model compound (Fig. 5).61 The ECL emission of N-doped TiO2 NTs can be sensitized by CdS QDs and quenched by CdTe QDs, which have been applied as strategies for the detection of adenosine in cancer cells139 and prostate protein antigen (PSA) in serum.140 Other MOS ECL nanoemitters include CdS@ZnO,93 ZnO nanoflowers,141 ZnO nanospheres, ZnS@ZnO and ZnSe@ZnO core–shell nanostructures,142 8-hydroxyquinoline aluminium nanoflowers,143etc. They have potential ECL applications as non-toxic emitters. The phosphopeptides can specifically adsorb onto the MOS nanomaterials, suggesting a plausible approach to biofunctionalize these materials for more ECL applications. Unannealed or uncalcinated MOS NPs normally possess a good deal of surface defects which facilitate the adsorption and entrapment of ECL coreactants, such as O2, H2O2 and K2S2O8.22,61 It is reported that a trace amount of O2 adsorbed onto TiO2 NTs before deaerization can hold the ECL intensity. The defects energetically correspond to metastable states, resulting in excited states with long lifetime and a unique high background signal of CL after the ECL emission, which is especially obvious for TiO2 (ref. 19 and 22) and CeO2–TiO2 composites.21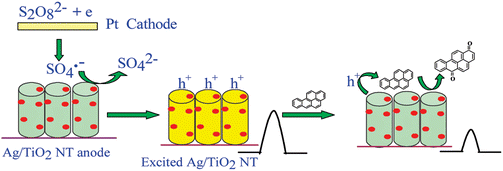 | ||
| Fig. 5 Schematic showing the ECL detection of PAHs with Ag/TiO2 NTs in S2O82− solution. (Reproduced from ref. 61. Copyright 2010, American Chemical Society.) | ||
5.6. Upconverting nanoparticles
The ECL nanoemitters mentioned above normally have PL emission with a Stokes shift. By contrast, the ECL of upconverting NPs has the PL of an anti-Stokes shift.144 A graphene-upconversion nanocomposite has been prepared with a one-step in situ hydrothermal method. The ECL intensity of NaYF4:Yb3+,Er3+ NPs is significantly amplified by graphene (Fig. 6).145 The lanthanides-doped NPs emit downconverting ECL while upconverting PL. An authentic electrogenerated upconverted emission has been validated from Ru(bpy)32+-doped 9,10-diphenylanthracene (DPA) nanowires.146 Conventional QDs and elemental NCs also have rare upconverting PL with special spectral terms corresponding to hyperfine structure transitions.144,147 From this point, NPs-based ECL is a generalized or superposed luminescence which can be fractionized into electro-chemi-fluorescence, electro-chemi-phosphorescence122–129 and electro-chemi-upconversion.145,146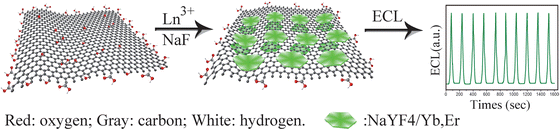 | ||
| Fig. 6 Schematic representation of preparation procedure of the graphene-NaYF4/Yb,Er nanohybrid and its ECL property. (Reproduced from ref. 145. Copyright 2012, Royal Society of Chemistry.) | ||
5.7. Molecular nanoaggregates
The conventional molecular ECL emitters like PAHs and polymers can be assembled into nanoscale aggregates which can still restore their ECL capability and even gain improvement. For instance, the ECL of 9,10-diphenylanthracene nanorods has been reported.24–26,146 The electrogenerated upconversion has been achieved in the uniformly doped organic nanowires based on triplet energy transfer from Ru(bpy)32+ to DPA.146 The ECL behaviors of various ruthenium bipyridyl (bpy) metallopolymer nanocomposites, such as [Ru(bpy)2(PVP)10]n(ClO4)2,148 [PVP-Ru(bpy)2Cl−]+/2+149 and [Ru(bpy)2(PVP)10Os(bpy)2]4+,150 have been investigated (PVP is short for poly(4-vinylpyridine)). Their electron transfer kinetics were further systematically discussed by the incorporation of AuNPs148 and the formation of inherently conducting polymers-composited hydrogel films.149 The visualized colorful ECL emission can be observed from poly(9,9-dioctylfluorene-co-benzothiadiazole) block copolymer NPs at the single particle level (Fig. 7)151 and patterned electrode,152 respectively. Because of the high sensitivity, low background and spatiotemporal coupled potential resolution, this visualized ECL prototype is very promising to open a broad avenue for multidimensional and multiplexed ECL imaging analyses.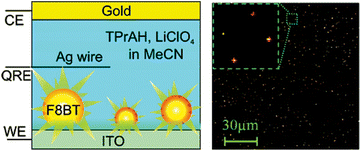 | ||
| Fig. 7 Schematic diagram of single molecule spectroelectrochemistry cell and wide-field ECL image with the laser beam focused on the central area of the image (inset: expanded region showing ECL from four particles). (Reproduced from ref. 151. Copyright 2008, American Chemical Society.) | ||
5.8. Hierarchical assembly of nanoemitters
Just like the ECL from the molecular aggregations, transmogrification of ECL nanoemitters into hierarchical architecture is also a practicable approach.60,89,102,106,116–120,153–156 The stacking mode of individual NPs may lead to an important effect on their ECL sensing applications. For example, hierarchical CdS nanotube arrays with high porosity and uniform alignment display considerably enhanced solid-state ECL in H2O2 solution compared with those of random aggregates.60,102 The assembly of QDs into nanospheres,115,116,155 hollow spheres,155 nanotubes,60,89,106,117 nanobelts,89 nanoflakes,119,120 and dendritic118 morphologies has also been reported with the demonstration of their ECL properties. These works not only greatly extend the ECL systems from quasi-zero nanodots to multidimensional nanomaterials and even microscaled block materials,155,156 but also provide more biofunctionalized nanoemitters for PL application.157From the aspect of electrons and orbitals, every PL emitter may acquire its ECL emission because the only distinction between PL and ECL lies at the stimulating source. It is always a robust field to find novel ECL nanoemitters by exhaustive searching among nanomaterials originally designed for optical applications, screening them with the ECL fundamentals, and then examining their ECL capabilities. For ECL bioanalysis, further functionalization of various nanoemitters with multitudinous biomolecules through physical adsorption, electrostatic interaction, covalent binding and specific affinity interaction has attracted considerable interest for building excellent ECL signal-transduction platforms.7,8,51,52
6. Improvement in ECL performance
Two major drawbacks are shared in NPs-based ECL systems: a high emission potential and low ECL intensity.66,78 Most reported cathodic ECL phenomena require a rather negative driven potential (e.g. below −1.0 V).53,82,85,87–96 The high applying potential may lead to undesirable side reactions in subsequent fabrications.In sensing procedures, an enhanced ECL intensity is preferred for their high sensitivity and detection performance. However, the intensity of NPs-based ECL is normally not at the level of Ru(bpy)32+ and luminol.7,8,52 Although instrumental parameters like the scan rate and PMT bias can be operationally modulated to partially realize intensive ECL emission,45,47,53,78,82,87–90 they also unexpectedly increase the background noise. General viable alternatives include the modulation of synthetic conditions of NPs,47,50,112 condensation of NP emitters,44,76,86,99 optimization of electrolytes and solution pH,40,47,50,76,99,112 and choice of specific bulk working electrode.50,112 However, these empirical implements still only bring limited progress.
To achieve efficient ECL emission from QDs, the introduction of an exogenous strong coreactant at an appropriate concentration is a seemingly most direct way, especially for anodic ECL. For example, the addition of SO32− can greatly enhance the ECL emission and thus improve the sensitivity for the ECL detection of dopamine50,109 nitrite,110 and tyrosine.112 However, coreactants cannot decrease the ECL potential. By contrast, some coreactants increase the potential.44,53,86,88,89,95,96 Modification of the NPs-based ECL system with functional materials has been taken as a conventional amplification strategy for fabricating novel and sensitive biosensors.64,66,73–78
6.1. Carbon nanomaterials
Several carbon nanomaterials with different nano-dimensions have been applied in improving the ECL behaviors of NPs, such as carbon nanotubes (CNTs),87,89,92,103,107,108,140,141,159,161,164,165 carbon nanospheres (CNSs),100 carbon nanofibers,158 and graphene90,98,105,113,131,145,160,166–168etc. These nanomaterials can not only be simply taken as modification substrates promoting the electron transfer between the immobilized NPs and the electrode, but can also be used as nanocarriers to assemble multiple NPs by electrostatic interaction mediated with polyelectrolytes due to their large specific areas, which is very suitable for labeling as tracing biotags.44,113,140 For example, the ECL of CdSe QDs can be greatly enhanced by the combination of CNTs and PDDA in CdSe QDs film,75 and CNSs with uniform size can greatly enhance the ECL of CdS QDs.100 K+-doped graphene by π-conjugation of K+-chelators onto the graphene basal plane has been demonstrated as a charge transfer accelerator for promoting the ECL emission of SiO2@CdS QDs nanocomposites.166 The oxygenated species by acid treatment on the edge and basal planes of nanocarbons can greatly favor the adsorption of the dissolved oxygen as a coreactant, which facilitates its conversion into radicals44,113 and results in a different sensor architecture. By facilitating the oxidation of CdTe QDs and triggering O2˙− generation, GO can enhance the anodic ECL for sensing glutathione.113Furthermore, the in situ growth of NPs at carbon nanomaterials has been demonstrated as a more intimate way to integrate the two components into nanocomposites,105,107,131 such as CdS QDs@CNTs.107 Compared with pure CdS NPs, CdS QDs@CNTs can enhance the ECL intensity by 5.3-fold and move the onset ECL potential positively by about 400 mV, which reduces H2O2 decomposition at the electrode surface and increases the detection sensitivity of H2O2. Graphene, owing to its large surface area, is also employed as the template for the in situ growth of NPs such as CdS QDs@graphene105 and AuNCs@graphene.131 This preparation route may result in a wide size distribution and is currently restricted to a few kinds of NPs with easy formation.65,105,131,169,170 CNTs with dopants like N have especially shown an amazing improvement in NPs-based ECL. The cathodic ECL emission of CdSe QDs composited with N-doped CNTs is 5-times stronger than that from pure QDs, and 3-times stronger than that composited with undoped CNTs.103 Taking advantage of the intrinsically strong adsorption of O2 on N-doped CNTs, a ‘signal-on’ ECL immunoassay has been proposed by the adsorption-induced concentration of coreactant and signal amplification.44
Due to the so-called ‘blackbody effect’,105,107 carbon nanomaterials are effective luminescence quenchers through energy transfer.171 Oxygen-functionalities can improve the annihilation efficiency by transforming the local conformation of π-conjugated nanocarbons into PAHs.172–175 Therefore, the NPs-based ECL enhancement by carbon nanomaterials is actually a constant competence between electrochemically boosting the production of R* and antagonistically annihilating its irradiation optically.98,105,107 Considering the electro-reduction capability of graphene,98,176–178 fully reduced carbon nanomaterials with restored backbones are ideal and are recommended to be employed as substrates in NPs-based ECL systems.
6.2. Metallic particles
Taking advantage of their excellent conductivity, metallic NPs, especially gold (AuNPs)53,82,88,91,104,108 and silver (AgNPs),61,159,179 can also reduce the electron-relay barrier between semiconducting ECL emitters and the electrode, accelerating the electron-/hole-injecting rate, thus not only obviously enhancing the ECL intensity, but also moving the ECL onset and peak potential toward zero.AuNPs, as one of the most common NPs in analytical techniques, have shown multifunctionality in NPs-based ECL assays. Their prime utilization is to increase the ECL intensity of NPs. Meanwhile, they are adept at immobilizing target biomolecules in constructing ECL sensors. A CdS QD-based ECL immunosensor for low-density lipoprotein by increasing its sensitivity with AuNPs amplification has been developed.53 CdS@CdSe QDs are assembled upon AuNPs-encapsulated silica nanospheres for highly enhanced ECL detection of a protein tumor marker.82 The in situ electrodeposition of AgNPs into CdS hierarchical nanoarrays179 as well as vertically aligned TiO2 nanotubes61 as templates have been well-defined. AgNPs can greatly enhance the ECL emission of these nanomaterials. The enhanced ECL of the CdS– and TiO2–Ag nanocomposite arrays leads to two sensitive sensors for H2O2179 and benzopyrene.61
Metallic NPs can have another contrasting effect on NPs-based ECL. The proximal luminescence quenching induced by their surface plasma resonance (SPR) cannot be ignored, especially for solid-state ECL.104,167,180–182 This effect can be inhibited by optimizing the size and amount of AuNPs and modulating their morphological anisotropy.104 As a methodological adaptation of surface-enhanced Raman spectroscopy (SERS), the SPR-induced near-field ECL energy transfer enhancement is otherwise surprisingly useful by keeping certain distance between the NPs-based ECL emitters and metallic NPs,167,180–182 which also realizes different sensor architecture by changing the relative space position of metallic NPs and semiconducting NPs.
6.3. Semiconductor metallic oxides
Semiconductor metallic oxide or MOS can improve NP-based ECL emission by shortening the path of electron transport between the conduction band of the NPs and the HOMO of the coreactant,21,57 which is quite different from the role of the above two nanomaterials. With the high-level conduction band and wide band-gap, MOS like TiO2 can offer indirect-band-gap-conductive electron transfer. As for ECL, integrating QDs with TiO2 can generate a metastable state to accept electrons and transmit them to the coreactant. The transfer of electrons from reduced QDs to TiO2 is helpful to improve the QD recovery efficiency, which also benefits enhanced and stable light emission in the ECL process.58 As TiO2 NTs can produce ECL emission with an approximating band-gap with CdS QDs, QDs can also sensitize the ECL emission of TiO2 NTs.1406.4. Other molecules and ions
Despite their non-conductivity, polyelectrolytes like PDDA89 and chitosan67,87,108 possess excellent film-forming abilities, which improves the distribution of the immobilized NPs and protects NPs from leakage, leading to a high ECL intensity. The highly branched generation 5.0 polyamidoamine dendrimer can regulate the size of QDs encapsulated within its interior structure to promote their ECL behavior.183 Compared to bare QDs, the core–shell CdSe/ZnS QDs incorporated within a Nafion polymer film show enhanced ECL when using H2O2 as a coreactant, the permselective properties of which significantly and effectively excluded S2O82− as the coreactant.184 Room-temperature ionic liquids (RTILs), as good ionic conductors, can also greatly increase the ECL intensity of QDs in solution.185 Recently, ECL enhancement of CdTe QDs by the addition of Ag+ in phosphate–citric acid and phosphate buffer has been reported.186 The maximum enhancement factor is of about 4.7. Biosensing strategies and applications
Novel biosensing methodologies have now become the main driving force of innovation for studying NPs-based ECL. An NPs-based ECL biosensing system normally contains three essential elements: bioactive substances as the biometric recognition element, ECL nanoemitters as the energy transducer, and coreactants as the signal sensitizer.52,187 The development of NPs-based ECL methodologies is always related to these elements.7.1. ECL inhibition/enhancement
The inhibition/enhancement effect of the analytes on the ECL emission of nanoemitters leads to an analytical methodology. It is especially facile to detect and quantify small molecules and ions. The first report on the analytical application of QDs-based ECL was proposed by combining CdSe QDs with a carbon paste electrode to produce ECL emission in an aqueous system, which could be enhanced by H2O2, leading to a method for H2O2 detection.45 Although the net results are expressed mainly as the change in the ECL intensity, the inner interactions may be different. For example, freely diffused analyte or analyte correlate can directly approach and interact with NPs to produce ECL energy transfer42,50,109,112,185 or electron transfer,14,60,61,188 or grab ligands competitively ravaging the surface states of NPs,15,76,77,79,189 and react with coreactants;19,22,40,45,47,63,102–106,110,113,131,154,156,160,162,163,181,188,192 additionally, various analyte-concentrating procedures have been affiliated prior to the above proposed interactions, which intrinsically feature the subtle roles of ECL nanoemitters as well as the coreactants in specific biosensing systems. This pre-enrichment has been validated through multiple approaches such as the adsorption of coreactants,45,61,112 potential-driven adsorption,191 biocatalytic precipitation,192etc. Based on these mechanisms, several novel and tricky detection applications have been proposed.The processes mentioned above have led to different ECL methods for the detection of a wide series of analytes, such as O2,19,32,130 H2O2,22,45,63,102,103–106,131,154,156,158,161,179,190 organic amines,40,186 polyphenols,185 halides,42 non-protein thiols,47,78,112 porphyrin derivatives,20,160 catechol derivatives,14,50,109,111 amino acids,188 nitrocompounds,60,110 Pb2+,15 Cu2+,76,77,79,189 and Hg2+.76
As an application example of the ECL energy transfer mechanism, a method for dopamine detection is based on energy transfer from excited QDs to the electro-oxidized product of dopamine, which quenches the excited QDs, thus decreasing the ECL emission.50 Based on electron transfer between hemin as prosthetic group and O2, the ECL quencher hemoglobin (Hb) can be measured by inhibiting the transformation of O2 to H2O2 at TiO2 NTs. The quenched ECL emission follows the Stern–Volmer equation in a wide linear range with a low cathodic potential and acceptable sensitivity.20
Based on the competition of metal ions and the stabilizer, the quenching effects of Cu2+ on the ECL emission of DMSA-capped CdTe QDs76 and L-cysteine-capped NIR-emitting CdSeTe alloyed QDs79 have been utilized to realize Cu2+. The former shows a linear range from 5.0 nM to 7.0 μM with a detection limit of 3.0 nM.77 This method can be extended to detect other metal ions like Hg2+ with a stronger metal–S interaction than with the Cd–S bond.76 The ECL of bovine serum albumin-protected AuNCs can also be inhibited by Pb2+.15
GO was demonstrated to facilitate QDs oxidation and trigger the transformation of O2 with extra strong adsorption toward O2˙−, leading to a 5-fold anodic ECL amplification of CdTe QDs even in deaerated conditions.112 The as-prepared ECL platform was realized for the sensitive and selective detection of glutathione from thiol-containing compounds and was further used for glutathione drug detection. Similarly, a facile signal amplification strategy was developed for a ‘signal-on’ immunoassay based on a novel ECL mechanism of CdS QDs by the adsorption-induced catalytic reduction of dissolved O2 on N-doped CNTs,45 which produces O2˙− to enhance the ECL emission. The polyelectrolyte functionalized N-doped CNTs showed a nearly two-fold stronger adsorption ability towards dissolved O2 than CNTs, which leads to faster formation of O2˙− and thus improves the ECL emission of the immunosensor.45
The strategy of analyte-induced ECL inhibition or enhancement can be implemented either in a homogeneous system of which the ECL nanoemitters are to be dissolved with some solute together with the coreactant and the analyte;15,40,42,47,50,78,109,110,185,188–190 or in a solid-state fashion in which the nanoemitters are immobilized on the electrode with or without coupling agents or spacers. The former ‘one-pot’ system is relatively facile to fabricate. However, the limited diffusion rate of every component and the low-solubility ECL nanoemitters give low intensity. More importantly, the entire mixture is susceptible to interferences, especially from real samples, and cannot be regenerated. Due to the direct contact with the electrode that ensures a direct and fast pathway for charge transfer, solid-state NPs-based ECL attains more efficient and stable electrochemistry and ECL signals than the homogeneous system.43,45 Moreover, the immobilized NPs as a biosensing substrate can further serve as an interface for stepwise modification and biorecognition.52
7.2. Steric hindrance from biorecognition reaction
Due to the insulation of biomacromolecules, the recognition of the biosensing interface for these molecules leads to steric hindrance, which not only impedes the mass transfer of the coreactant, but also decelerates the electron transfer among the NPs, electrode and coreactant, thus lowering the ECL intensity. This strategy is mainly applied for label-free immunoassays.45,53,82,87–90,166,194,195 For example, a label-free ECL biosensor for low-density lipoprotein (LDL) has been developed.53 The ECL intensity decreases linearly with the increasing LDL concentration from 0.025 to 16 ng mL−1. Based on the co-immobilization of antibodies with CdSe QDs/CNTs-chitosan on the electrode surface and the increasing resistance upon formation of the immunocomplex between the antibody and the target antigen, a label-free ECL immunoassay strategy has been developed for the sensitive detection of human IgG (HIgG).87 Another ECL immunosensor based on amplification of polyelectrolyte-protected graphene shows a highly sensitive response to HIgG in the linear range of 0.02–2000 pg mL−1 with an LOD of 0.005 pg mL−1 (Fig. 8).90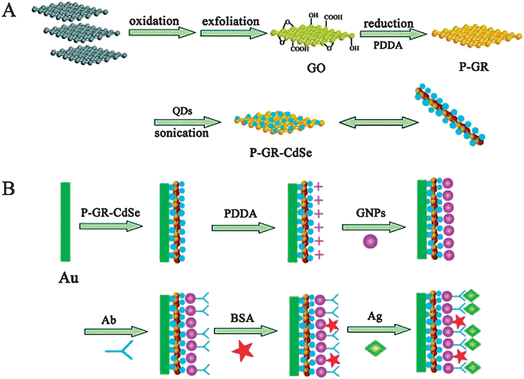 | ||
| Fig. 8 (A) Schematic representation of preparation procedure of P-GR-CdSe composites, including the oxidation of graphite (gray blocks) to GO with abundant oxygen functionalities, the in situ reduction of GO in the presence of PDDA to obtain positively charged PDDA protected graphene (P-GR) colloids, and the preparation of P-GR-CdSe composites via electrostatic interactions under sonication. (B) Schematic illustration of the stepwise immunosensor fabrication process, including the formation of the P-GR-CdSe composite film on the Au electrode, the linkage of PDDA to the film, the conjugation of gold NPs (GNPs) to PDDA, the immobilization of antibody (Ab) on the electrode via GNPs, and the specific immunoreaction. (Reproduced from ref. 90. Copyright 2011, Wiley InterScience.) | ||
This strategy is also suitable for cytosensing. An ECL cytosensor has been designed to analyze cell-membrane carbohydrates by combining the QDs-based ECL with specific recognition of lectins for carbohydrates.91 The specific recognition of QD-bound lectins for cell-surface carbohydrates can capture cells onto the electrode surface and thus decreases the ECL intensity which is proportional to the amount of captured cells. The ECL change thus provides a simple but highly sensitive method for cytosensing and the dynamic monitoring of cell-surface carbohydrate expression. By integrating the competitive recognition of lectin for cell-surface carbohydrate with a nanocomposited carbohydrate-functionalized CdS QDs-modified electrode, a similar ECL strategy has also been proposed for in situ, label-free monitoring of carbohydrate expression on living cells.92 The combination of lectin with an electrode surface leads to steric hindrance to the electron transfer, thus decreasing the ECL emission of the QDs.
7.3. Generation/consumption of coreactant
This methodology is mainly based on the generation of H2O2 or the consumption of dissolved oxygen in enzymatic reactions, which acts as the coreactant in NPs-based ECL reactions. It started with a QDs-based enzymatic sensor for glucose detection,43 in which CdSe QDs were co-immobilized with glucose oxidase (GOx). Upon addition of glucose, the oxidation of glucose catalyzed by GOx consumed dissolved oxygen, thus decreasing the ECL response. A disposable CdTe QDs-based ECL glucose biosensor has also been developed on a screen-printed carbon paste electrode (SPCE).99 This strategy can be extended to more systems for the detection of oxidase substrates, such as acetylcholine and choline98 and hypoxanthine.100The enzymatic reaction can coupled with the analyte-inhibition/enhancement strategy to achieve a dramatic amplification in the ECL signal by an enzymatic catalytic cycle. For example, the electro-oxidized product of tyrosine can quench the excited MPA-capped CdTe QDs. In order to further increase the sensitivity, tyrosinase is used to catalyze the oxidation of tyrosine by dissolved O2 to produce the quencher o-quinone, resulting in a sub-picomolar LOD for tyrosine.50 A similar method for the detection of catechol derivatives has also been developed by the cathodic ECL emission of DMSA-stabilized CdTe QDs with low-cost horseradish peroxidase (HRP).66
Due to the high signal stability of the multilayer film and excellent electrocatalytical ability of Hb toward H2O2, a ‘signal-on’ ECL sensor for the detection of H2O2 has been developed using CdS QDs and an Hb multilayer film {Hb/CdS}n.193 Another ‘signal-on’ ECL bienzyme biosensor is constructed by the in situ formation of CdS QDs on multi-walled CNTs (MWCNTs).107 This sensor shows wide linear ranges with detection limits of 0.8 and 1.7 mM for the detection of choline and acetylcholine, respectively.
The consumption of the ECL coreactant resulting from enzymatic reactions can be triggered by enzyme-labeled antibodies, which enables NPs-based ECL immunosensing. For example, a sensitive and competitive immunosensor based on the ECL of DMSA-stabilized CdTe QDs has been proposed.67 Upon immuno-recognition of the immobilized antigen for the HRP-labeled antibody, the ECL intensity decreases as an enzymatic reaction consumes the self-produced coreactant in the presence of hydroquinone, leading to a wide calibration range of 0.05 ng mL−1 to 5 μg mL−1 for HIgG. The enzyme-labeled antibody can be further condensed on nanocarriers to achieve a higher tagging ratio and a more sensitive performance.
As enzyme-linked antibodies usually suffer from needing a specific catalytic substrate, costly engineering and purification process, and the easy loss of bioactivity,86,108 the non-enzymatic tag is becoming an alternative for NPs-based ECL immunoassay. For example, a DNAzyme-labeled antibody has been designed by assembling an antibody and hemin aptamer on AuNPs to form a G-quadruplex/hemin bio-barcode. Owing to the consumption of oxygen by a DNAzyme-catalyzed reaction, the QDs-based ECL is quenched upon the formation of the immunocomplex, leading to a linear range of 0.01 pg mL−1 to 1 ng mL−1 for the sensitive immunoassay of α-fetoprotein.108 Another ECL immunoassay method by using a dendrimer-encapsulated PdNPs-anchored carbon nanohorn as a non-enzymatic tracing tag to electrocatalytically reduce dissolved oxygen has also been proposed. The consumption of dissolved oxygen as the coreactant leads to a decrease of the ECL emission in a linear concentration range of carcinoembryonic antigen over six orders of magnitude (Fig. 9).86
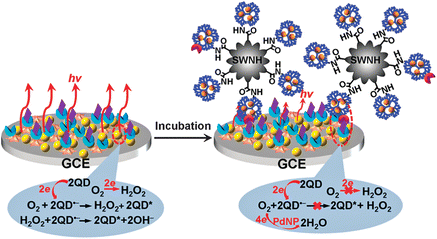 | ||
| Fig. 9 Schematic representation of preparations of tracing tag, and ECL annihilation strategy by electrocatalytic reduction towards dissolved O2 at PdNPs@generation-5.0 dendrimer/single-walled nanohorn nanohybrids. (Reproduced from ref. 86. Copyright 2012, Royal Society of Chemistry.) | ||
7.4. ECL resonance energy transfer
ECL resonance energy transfer (ERET) can happen at a relative long distance in comparison with FRET.123 This strategy emphasizes the ERET between ECL nanoemitters and the probes or tracing tags for immuno- and DNA-assays.64,93,123,139,140,180–182 It does not require the additional introduction of any free molecule as the donor or acceptor, and thus can be taken as a reagentless strategy.An application example is based on the energy transfer from the electrochemically excited Mn2+-doped CdS QDs to AuNPs to quench the ECL emission.104 The AuNPs are used to label a DNA probe. After hybridization with the target DNA, the distance between the AuNPs and QDs is elongated, leading to the enhancement of the ECL emission.64 By swapping the positions of the undoped CdS QDs and AuNPs in this framework, a similar aptasensor has been proposed in sandwich format.181 Another ultrasensitive DNA detection approach has been reported by combining AuNP-based ERET with an isothermal circular amplification reaction, which can sensitively respond to DNA down to 5 aM (Fig. 10).182
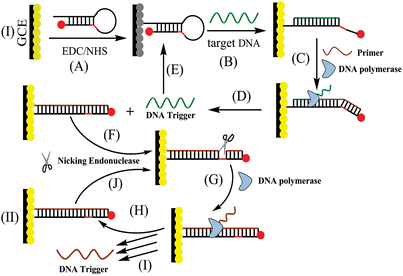 | ||
| Fig. 10 Schematic representation of AuNPs and isothermal circular assisted ECL signal amplification of the CdS QDs film for sensitive assay of nucleic acids. (Reproduced from ref. 182. Copyright 2011, Royal Society of Chemistry.) | ||
The ECL emission of Mn2+-doped CdS QDs can stir the magnetic field of superparamagnetic Fe3O4 NPs by changing their spin multiplicity, accompanied by a feedback of electromagnetic induction in the QDs. This opto-magnetic interaction can induce an enhancement in theECL intensity. Using the Fe3O4 NPs-labeled signal antibody as a probe, a ‘signal-on’ QDs-based ECL sandwich-type immunoassay has been developed for ultrasensitive antigen detection.122
Surface carboxyls-activated CdTe QDs-based ECL can effectively scavenge the ECL energy of Mn2+-doped CdS QDs. Thus combining Mn2+-doped CdS QDs and antibody co-immobilized electrode with a CdTe QD labeled antibody, an ultrasensitive immunosensor for mouse IgG is proposed.177 A similar work has also demonstrated that the quenching of ECL emission from an Mn2+-doped CdS QDs film by CdTe QDs via an energy scavenging process can be amplified by the incorporation of a large number of QDs into the silica matrix for DNA detection.64 Furthermore, a facile signal amplification strategy for the sensitive and quantitative detection of β2M expressed SMMC-7721 cells has been proposed based on ERET between an antibody-modified CdS QDs donor and a Ru(bpy)32+ acceptor labeled on a cell surface.93 In this system, CdS QDs can be substituted by other ECL nanoemitters like TiO2 NTs.140 Combined with the newly advent optical nanomaterials such as upconverting NPs,144 metallic NCs,13–15,131–133 and carbon nanodots,18 this methodology will be further developed.
7.5. Nanoemitters as labels for ECL detection
ECL nanoemitters can be either dissolved in a homogenous phase or immobilized in the solid state for analytical applications. When nanoemitters are used in the solid state, they can be further divided into three groups: (1) entrapped in the electrode; (2) immobilized as a sensing substrate at the electrode surface; and (3) labeled on recognition molecules. In the earlier NPs-based ECL studies, the entrapment of NPs has presented a possible way mainly for the investigation of mechanisms such as CdSe QDs in a graphite-incubated paraffin electrode45 and CdS NTs in a carbon paste electrode.106 Later, the formation of an NP film on an electrode surface received much attention, where the film can be regenerated and used repeatedly. To make the best of the high sensitivity of ECL techniques, developing nanoemitters-labeled ECL analytical methods is self-evident, as these have barely any background and normally show a ‘turn-on’ response. NPs as ECL emitters also enable more versatile ways for preparing signal tags than the majority of Ru(bpy)32+-doped SiO2 NPs196 and luminol-doped AuNPs.197 By employing nanoemitter-labeled biomolecules, the NPs-based ECL methodology is endowed with high specific biorecognition, such as antibody–antigen recognition for immunoassays,41,85,95,96,156 nucleotide duplex interaction for DNA analyses,83,159,179,198–200 and aptamer–target association for aptasensing.164,165,198,199,201 Moreover, this methodology can easily be integrated with magnetic separation and enrichment,83–85,94,159 meeting the demand for automated, high-throughput, multiplexed analysis, thus promising the prospect of commercialization. Nevertheless, steric hindrance grows obviously in this strategy, which could be relieved by the above-mentioned ECL-improvement implementations. This strategy was initiated with the QDs-based ECL immunoassay41,85,95,96,156,165 and now enjoys exceptional advantages in DNA detection as well as far much less blocking of charge transfer in single- or double-stranded chains.83,160,164,185,196–199 Hence, it can be designated as a nanoemitter-labeled ECL strategy in which the ECL intensity correlates to the amount of labels.An ECL immunosensor using CdTe QDs-coated SiO2 NPs as labels has been proposed for the ultrasensitive detection of a biomarker.95 Due to the signal amplification from the high loading of CdTe QDs, a 6.6-fold enhancement in the ECL signal for IgG detection is achieved compared to the unamplified method. An ECL signal amplification strategy for the sensitive detection of tumor necrosis factor-alpha using QDs-polymer-functionalized SiO2 NPs as the label has also been proposed (Fig. 11).96 An ultrasensitive ECL immunosensor has been developed using PtAg@CNCs as excellent labels based on CNTs-chitosan/AuNPs composite-modified SPCEs for PSA detection.156 The core–shell ZnS@CdSe QDs coated with a carboxyl polymer layer is used as an ECL label for a sandwich-type immunoassay of C-reactive protein at Au rotating disk electrodes with DBAE as the coreactant. The limit of detection is 1.0 mg mL−1,41 which is the only NP-based anodic ECL immunoassay up to now.
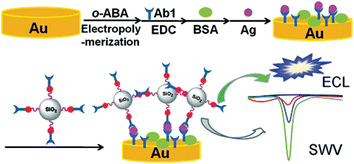 | ||
| Fig. 11 Schematic representation of the sandwich immunoassay with Si/PGMA/QD/Ab2 as the label. (Reproduced from ref. 96. Copyright 2011, American Chemical Society.) | ||
A biosensor for thrombin detection has been reported in a QD-labeled technique. The thiol-terminated aptamer was first immobilized on an Au electrode, and then thrombin was imported to form the aptamer–thrombin bioaffinity complex. Another 5′-biotin-modified aptamer was next hybridized with the combined thrombin to form a sandwich-type structure following the binding of streptavidin-modified QDs via the biotin–avidin specific recognition.196 A facile strategy for the fabrication of the aptamer-based adenosine 5′-triphosphate biosensor has also been developed in a similar way.199 A QDs-labeled ECL biosensor for the detection of lysozyme has been developed by forming the aptamer–lysozyme bioaffinity complexes at an Au electrode. The free probes are hybridized with the 5′-biotin-modified complementary oligonucleotides to form double-stranded oligonucleotides. Avidin-QDs are bound through the biotin–avidin-system. The ECL signal is responsive to the amount of QDs, which is indirectly inversely proportional to the combined target protein.197
The dendrimer/ZnS@CdSe QDs NC has been fabricated and used as an ECL probe combined with magnetic beads (MBs) for aptamer immobilization for the signal-on ECL assay of cancer cells (Fig. 12).85 A cycle-amplifying technique using a DNA device on MBs was further employed in the ECL assay of cancer cells, which greatly improves the sensitivity.85 Also, an ultrasensitive ECL biosensor has been developed for the detection of near single DNA molecules with a linear range of seven orders of magnitude by combining the specific recognition of a molecular beacon with the signal amplification of QDs-dendrimer nanocomposites.183 Another sensitive strategy which integrates a DNA cycle device onto MBs amplifies the signal with GO and enhances the ECL intensity of probe DNA-labeled CdS QDs as nicking product which has been developed for thrombin detection.159
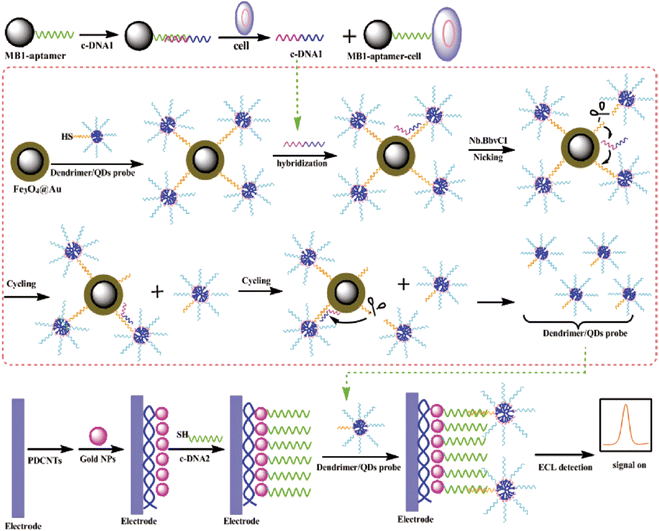 | ||
| Fig. 12 Schematic representation of the strategy for cell assay based on the MB–aptamer biocomplex and DNA cycle-amplifying technique and ECL detection based on the dendrimer NCs/QDs-DNA signal probe. (Reproduced from ref. 85. Copyright 2011, American Chemical Society.) | ||
8. Prospective
To meet the nanomanufacturing demand for optoelectronic devices, fabricating solid-state disposable ECL paper-based and lab-on-chip platforms should be a priority for low-cost mass production and multiplexed point-of-care tests.99,202–204 Meanwhile, it is quite urgent to realize visible NP-based ECL electrochromics205 and imaging technology151,152,206–216 for spatiotemporally-resolved multiplexed analysis and highly selective single-molecule detection in biological systems without PMT amplification. Therefore, highly efficient, low applied potential and tunable NP-based ECL systems (emitters and coreactants) may receive major efforts. The biocompatible NIR-ECL nanoemitters and alloyed NCs as well as low-toxicity and eco-friendly carbon nanodots may take the lead as good candidates for the next generation nanoemitters, especially for preparing novel functional ECL probes, whereas doping, compositing or assembling with ferromagnetic elements or materials can bring into being multifunctional ECL nanoemitters or nanostructures for high-throughput magnetic separation and ECL-magnetic resonance imaging.According to the Interactive Link Maps of the International Union of Purified and Applied Chemistry (IUPAC), the node entry of ECL is extensively connected with other luminescent phenomena.217 Meanwhile, given the versatile physicochemical applications of NPs as nanoemitters, such as in PL, CL, SERS, spectroelectrochemistry, electroluminescence, cathodoluminescence, even electrochemistry, giant magnetoresistance, etc., these techniques may be digested, associated and adapted to inspire innovative ECL of NPs systems.
Furthermore, besides the biological amplification strategies mentioned above, DNA amplification techniques such as rolling circle amplification, target-induced repeated primer extension, hybridization chain reaction, loop-mediated amplification, and target recycling amplification with endonuclease, exonuclease and polymerase-based circular strand-replacement polymerization can be adopted to amplify the NPs-based ECL signal for the biosensing of proteins and nucleic acids.218
9. Conclusion
As a new family of ECL emitters, NPs have provided an entirely different sensing paradigm. Their functionalization with multitudinous biomolecules offer excellent platforms for the ultrasensitive quantification of a wide range of analytes through ECL signal transduction. It is a persistent research motivation to seek for novel, efficient NPs-based ECL systems (emitters and coreactants) combined with deep elucidation of their mechanisms and modern analytical techniques. The long-term needs in biomedicine, clinical diagnosis, food and environmental monitoring also continuously push the development of NPs-based ECL technology forwards.Acknowledgements
This work was financially supported by the National Basic Research Program of China (2010CB732400), National Natural Science Foundation of China (21121091, 21135002).References
- K. A. Fahnrich, M. Pravda and G. G. Guilbault, Talanta, 2001, 54, 531–559 CrossRef CAS.
- R. T. Dufford, D. Nightingale and L. W. Gaddum, J. Am. Chem. Soc., 1927, 49, 1858–1864 CrossRef CAS.
- N. Harvey, J. Phys. Chem., 1929, 33, 1456–1459 CrossRef CAS.
- A. Zweig, A. H. Maurer and B. G. Roberts, J. Org. Chem., 1967, 32, 1322–1329 CrossRef CAS.
- A. Zweig, D. L. Maricle, J. S. Brinen and A. H. Maurer, J. Am. Chem. Soc., 1967, 89, 473–474 CrossRef CAS.
- N. Tokel and A. J. Bard, J. Am. Chem. Soc., 1972, 94, 2862–2863 CrossRef CAS.
- M. M. Richter, Chem. Rev., 2004, 104, 3003–3036 CrossRef CAS.
- W. J. Miao, Chem. Rev., 2008, 108, 2506–2553 CrossRef CAS.
- Z. Ding, B. M. Quinn, S. K. Haram, L. E. Pell, B. A. Korgel and A. J. Bard, Science, 2002, 296, 1293–1297 CrossRef CAS.
- M. Bruchez, M. Moronne, P. Gin, S. Weiss and A. P. Alivisatos, Science, 1998, 281, 2013–2016 CrossRef CAS.
- W. C. W. Chan and S. M. Nie, Science, 1998, 281, 2016–2018 CrossRef CAS.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundarensan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS.
- I. Díez, M. Pusa, S. Kulmala, H. Jiang, A. Walther, A. S. Goldmann, A. H. Müller, O. Ikkala and R. H. Ras, Angew. Chem., Int. Ed., 2009, 48, 2122–2125 CrossRef.
- L. L. Li, Y. H. Liu, Y. Y. Shen, J. R. Zhang and J. J. Zhu, Anal. Chem., 2011, 83, 661–665 CrossRef CAS.
- Y. M. Fang, J. Song, J. Li, Y. W. Wang, H. H. Yang, J. J. Sun and G. N. Chen, Chem. Commun., 2011, 47, 2369–2371 RSC.
- L. Y. Zheng, Y. W. Chi, Y. Q. Dong, J. P. Lin and B. B. Wang, J. Am. Chem. Soc., 2009, 131, 4564–4565 CrossRef CAS.
- H. Zhu, X. L. Wang, Y. L. Li, Z. J. Wang, F. Yang and X. R. Yang, Chem. Commun., 2009, 5118–5120 RSC.
- S. N. Baker and G. A. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726–6744 CrossRef CAS.
- Z. Y. Lin, Y. Liu and G. N. Chen, Electrochem. Commun., 2008, 10, 1629–1632 CrossRef CAS.
- X. Liu, Z. T. Hou, J. P. Lei, H. X. Ju, J. X. Cai and J. Y. Shen, Electroanalysis, 2011, 23, 2629–2632 CrossRef CAS.
- Y. Zhu, G. Li, S. Y. Zhang, J. M. Song, C. J. Mao, H. L. Niu, B. K. Jin and Y. P. Tian, Electrochim. Acta, 2011, 56, 7550–7554 CrossRef CAS.
- L. F. Chen, L. L. Lu, Y. Mo, Z. M. Xu, S. P. Xie, H. Y. Yuan, D. Xiao and M. M. Choi, Talanta, 2011, 85, 56–62 CrossRef CAS.
- A. Devadoss, C. Dickinson, T. E. Keyes and R. J. Forster, Anal. Chem., 2011, 83, 2383–2387 CrossRef CAS.
- W. I. Lee, Y. Bae and A. J. Bard, J. Am. Chem. Soc., 2004, 126, 8358–8359 CrossRef CAS.
- Y. L. Chang, R. E. Palacios, F. R. F. Fan, A. J. Bard and P. F. Barbara, J. Am. Chem. Soc., 2008, 130, 8906–8907 CrossRef CAS.
- K. M. Omer and A. J. Bard, J. Phys. Chem. C, 2009, 113, 11575–11578 CAS.
- N. Myung, Z. Ding and A. J. Bard, Nano Lett., 2002, 2, 1315–1319 CrossRef CAS.
- N. Myung, Y. J. Bae and A. J. Bard, Nano Lett., 2003, 3, 1053–1055 CrossRef CAS.
- Y. J. Bae, N. Myung and A. J. Bard, Nano Lett., 2004, 4, 1153–1161 CrossRef CAS.
- N. Myung, X. Lu, K. P. Johnston and A. J. Bard, Nano Lett., 2004, 4, 183–185 CrossRef CAS.
- C. Lu, X. F. Wang, J. J. Xu and H. Y. Chen, Electrochem. Commun., 2008, 10, 1530–1532 CrossRef CAS.
- R. J. Zheng, Y. M. Fang, S. F. Qin, J. Song, A. H. Wu and J. J. Sun, Sens. Actuators, B, 2011, 157, 488–493 CrossRef CAS.
- L. Sun, L. Bao, B. R. Hyun, A. C. Bartnik, Y. W. Zhong, J. C. Reed, D. W. Pang, H. D. Abruña, G. G. Malliaras and F. W. Wise, Nano Lett., 2009, 9, 789–793 CrossRef CAS.
- S. K. Haram, B. M. Quinn and A. J. Bard, J. Am. Chem. Soc., 2001, 123, 8860–8861 CrossRef CAS.
- S. N. Inamdar, P. P. Ingole and S. K. Haram, ChemPhysChem, 2008, 9, 2574–2579 CrossRef CAS.
- S. K. Haram, A. Kshirsagar, Y. D. Gujarathi, P. P. Ingole, O. A. Nene, G. B. Markad and S. P. Nanavati, J. Phys. Chem. C, 2011, 115, 6243–6249 CAS.
- E. Talgorn, R. D. Abellon, P. J. Kooyman, J. Piris, T. J. Savenije, A. Goossens, A. J. Houtepen and L. D. A. Siebbeles, ACS Nano, 2010, 4, 1723–1731 CrossRef CAS.
- K. S. V. Santhanam and A. J. Bard, J. Am. Chem. Soc., 1965, 87, 139–140 CrossRef CAS.
- P. J. Smith and C. K. Mann, J. Org. Chem., 1969, 34, 1821–1826 CrossRef CAS.
- L. H. Zhang, X. Q. Zou, E. B. Ying and S. J. Dong, J. Phys. Chem. C, 2008, 112, 4451–4454 CAS.
- S. J. Wang, E. Harris, J. Shi, A. Chen, S. Parajuli, X. H. Jing and W. J. Miao, Phys. Chem. Chem. Phys., 2010, 12, 10073–10080 RSC.
- H. Jiang and X. M. Wang, Electrochem. Commun., 2009, 11, 1207–1210 CrossRef CAS.
- H. Jiang and H. Ju, Chem. Commun., 2007, 404–406 RSC.
- S. Y. Deng, Z. T. Hou, J. P. Lei, D. J. Lin, Z. Hu, F. Yan and H. X. Ju, Chem. Commun., 2011, 47, 12107–12109 RSC.
- G. Z. Zou and H. X. Ju, Anal. Chem., 2004, 76, 6871–6876 CrossRef CAS.
- J. P. Choi and A. J. Bard, Anal. Chim. Acta, 2005, 541, 141–148 CrossRef.
- H. Jiang and H. X. Ju, Anal. Chem., 2007, 79, 6690–6696 CrossRef CAS.
- H. S. White and A. J. Bard, J. Am. Chem. Soc., 1982, 104, 6891–6895 CrossRef CAS.
- M. M. Chang, T. Saji and A. J. Bard, J. Am. Chem. Soc., 1977, 99, 5399–5403 CrossRef CAS.
- X. Liu and H. X. Ju, Anal. Chem., 2008, 80, 5377–5382 CrossRef CAS.
- R. Gill, M. Zayats and I. Willner, Angew. Chem., Int. Ed., 2008, 47, 7602–7625 CrossRef CAS.
- J. P. Lei and H. Ju, TrAC, Trends Anal. Chem., 2011, 30, 1351–1359 CrossRef CAS.
- G. F. Jie, B. Liu, H. C. Pan, J. J. Zhu and H. Y. Chen, Anal. Chem., 2007, 79, 5574–5581 CrossRef CAS.
- A. H. Wu, J. J. Sun, X. L. Su, Y. W. Lin, Z. B. Lin, H. H. Yang and G. N. Chen, Analyst, 2010, 135, 2309–2315 RSC.
- H. Niu, R. Yuan, Y. Q. Chai, L. Mao, Y. L. Yuan, Y. Zhuo, S. R. Yuan and X. Yang, Biosens. Bioelectron., 2011, 26, 3175–3180 CrossRef CAS.
- L. Y. Zheng, Y. W. Chi, B. B. Wang, L. M. Han and G. N. Chen, Chem. Commun., 2010, 46, 5734–5736 RSC.
- S. N. Ding, B. H. Gao, D. Shan, Y. M. Sun and S. Cosnier, Chem.–Eur. J., 2012, 18, 1595–1598 CrossRef CAS.
- F. R. F. Fan, S. Park, Y. W. Zhu, R. S. Ruoff and A. J. Bard, J. Am. Chem. Soc., 2009, 131, 937–939 CrossRef CAS.
- Y. M. Fang, J. Song, R. J. Zheng, Y.-M. Zeng and J. J. Sun, J. Phys. Chem. C, 2011, 115, 9117–9121 CAS.
- Y. M. Fang, J. J. Sun, A. H. Wu, X. L. Su and G. N. Chen, Langmuir, 2009, 25, 555–560 CrossRef CAS.
- J. X. Li, L. X. Yang, S. L. Luo, B. B. Chen, J. Li, H. L. Lin, Q. Y. Cai and S. Z. Yao, Anal. Chem., 2010, 82, 7357–7361 CrossRef CAS.
- Y. Zhu, G. Li, S. Y. Zhang, J. M. Song, C. J. Mao, H. L. Niu, B. K. Jin and Y. P. Tian, Electrochim. Acta, 2011, 56, 7550–7554 CrossRef CAS.
- X. F. Wang, J. J. Xu and H. Y. Chen, J. Phys. Chem. C, 2008, 112, 17581–17585 CAS.
- Y. Shan, J. J. Xu and H. Y. Chen, Nanoscale, 2011, 3, 2916–2923 RSC.
- A. N. Cao, Z. Liu, S. S. Chu, M. H. Wu, Z. M. Ye, Z. W. Cai, Y. L. Chang, S. F. Wang, Q. H. Gong and Y. F. Liu, Adv. Mater., 2010, 22, 103–106 CrossRef CAS.
- X. Liu, L. Cheng, J. Lei, H. Liu and H. Ju, Chem.–Eur. J., 2010, 16, 10764–10770 CrossRef CAS.
- X. Liu, Y. Y. Zhang, J. P. Lei, Y. D. Xue, L. X. Cheng and H. X. Ju, Anal. Chem., 2010, 82, 7351–7356 CrossRef CAS.
- K. Kawahara, K. Suzuki, Y. Ohko and T. Tatsuma, Phys. Chem. Chem. Phys., 2005, 7, 3851–3855 RSC.
- I. Robel, M. Kuno and P. V. Kamat, J. Am. Chem. Soc., 2007, 129, 4136–4137 CrossRef CAS.
- M. Bottrill and M. Green, Chem. Commun., 2011, 47, 7039–7050 RSC.
- C. W. Ge, M. Xu, J. Liu, J. P. Lei and H. X. Ju, Chem. Commun., 2008, 450–452 RSC.
- R. Freeman, T. Finder, L. Bahshi and I. Willner, Nano Lett., 2009, 9, 2073–2076 CrossRef CAS.
- R. Freeman and I. Willner, Nano Lett., 2009, 9, 322–326 CrossRef CAS.
- R. Gill, R. Freeman, J. P. Xu, I. Willner, S. Winograd, I. Shweky and U. Banin, J. Am. Chem. Soc., 2006, 128, 15376–15377 CrossRef CAS.
- I. L. Medintz, T. Pons, S. A. Trammell, A. F. Grimes, D. S. English, J. B. Blanco-Canosa, P. E. Dawson and H. Mattoussi, J. Am. Chem. Soc., 2008, 130, 16745–16756 CrossRef CAS.
- L. X. Cheng, X. Liu, J. P. Lei and H. X. Ju, Anal. Chem., 2010, 82, 3359–3364 CrossRef CAS.
- Y. L. Mei, H. S. Wang, Y. F. Li, Z. Y. Pan and W. L. Jia, Electroanalysis, 2010, 22, 155–160 CrossRef CAS.
- G. D. Liang, L. P. Shen, G. Z. Zou and X. L. Zhang, Chem.–Eur. J., 2011, 17, 10213–10215 CrossRef CAS.
- G. X. Liang, H. Y. Liu, J. R. Zhang and J. J. Zhu, Talanta, 2010, 80, 2172–2176 CrossRef CAS.
- G. X. Liang, L. L. Li, H. Y. Liu, J. R. Zhang, C. Burda and J. J. Zhu, Chem. Commun., 2010, 46, 2974–2976 RSC.
- J. Wang, X. C. Jiang, H. Y. Han and N. Li, Electrochem. Commun., 2011, 13, 359–362 CrossRef CAS.
- G. F. Jie, P. Liu and S. S. Zhang, Chem. Commun., 2010, 46, 1323–1325 RSC.
- G. F. Jie, L. Wang and S. S. Zhang, Chem.–Eur. J., 2011, 17, 641–648 CrossRef CAS.
- G. F. Jie, J. X. Yuan and J. Zhang, Biosens. Bioelectron., 2012, 31, 69–76 CrossRef CAS.
- G. F. Jie, L. Wang, J. X. Yuan and S. S. Zhang, Anal. Chem., 2011, 83, 3873–3880 CrossRef CAS.
- S. Y. Deng, J. P. Lei, Y. Huang, X. N. Yao, L. Ding and H. X. Ju, Chem. Commun., 2012, 48, 9159–9161 RSC.
- G. F. Jie, J. J. Zhang, D. C. Wang, C. Cheng, H. Y. Chen and J. J. Zhu, Anal. Chem., 2008, 80, 4033–4039 CrossRef CAS.
- G. Jie, H. Huang, X. Sun and J. J. Zhu, Biosens. Bioelectron., 2008, 23, 1896–1899 CrossRef CAS.
- G. Jie, L. Li, C. Chen, J. Xuan and J. J. Zhu, Biosens. Bioelectron., 2009, 24, 3352–3358 CrossRef CAS.
- L. L. Li, K. P. Liu, G. H. Yang, C. M. Wang, J. R. Zhang and J.-J. Zhu, Adv. Funct. Mater., 2011, 21, 869–878 CrossRef CAS.
- E. Han, L. Ding, H. Lian and H. Ju, Chem. Commun., 2010, 46, 5446–5448 RSC.
- E. Han, L. Ding, S. Jin and H. Ju, Biosens. Bioelectron., 2011, 26, 2500–2505 CrossRef CAS.
- M. S. Wu, H. W. Shi, J. J. Xu and H. Y. Chen, Chem. Commun., 2011, 47, 7752–7754 RSC.
- C. F. Ding, Y. Ge and S. S. Zhang, Chem.–Eur. J., 2010, 16, 10707–10714 CrossRef CAS.
- J. Qian, C. Y. Zhang, X. D. Cao and S. Q. Liu, Anal. Chem., 2010, 82, 6422–6429 CrossRef CAS.
- L. Yuan, X. Hua, Y. F. Wu, X. H. Pan and S. Q. Liu, Anal. Chem., 2011, 83, 6800–6809 CrossRef CAS.
- S. Maldonado and K. J. Stevenson, J. Phys. Chem. B, 2004, 108, 11375–11383 CrossRef CAS.
- S. Y. Deng, J. P. Lei, L. X. Cheng, Y. Y. Zhang and H. X. Ju, Biosens. Bioelectron., 2011, 26, 4552–4558 CrossRef CAS.
- L. X. Cheng, S. Y. Deng, J. P. Lei and H. X. Ju, Analyst, 2012, 137, 140–144 RSC.
- Y. Y. Zhang, S. Y. Deng, J. P. Lei, Q. N. Xu and H. X. Ju, Talanta, 2011, 85, 2154–2158 CrossRef CAS.
- B. Yue, Y. W. Ma, H. S. Tao, L. S. Yu, G. Q. Jian, X. Z. Wang, X. S. Wang, Y. N. Lu and Z. Hu, J. Mater. Chem., 2008, 18, 1–5 RSC.
- C. Z. Wang, E. F. Yifeng, L. Z. Fan, Z. H. Wang, H. B. Liu, Y. L. Li and S. H. Yang, Adv. Mater., 2007, 19, 3677–3681 CrossRef CAS.
- L. Guo, X. Liu, Z. Hu, S. Deng and H. Ju, Electroanalysis, 2009, 21, 2495–2498 CrossRef CAS.
- C. Shi, Y. Shan, J. Xu and H. Chen, Electrochim. Acta, 2010, 55, 8268–8272 CrossRef CAS.
- K. Wang, Q. Liu, X. Y. Wu, Q. M. Guan and H. N. Li, Talanta, 2010, 82, 372–376 CrossRef CAS.
- G. F. Jie, B. Liu, J. J. Miao and J. J. Zhu, Talanta, 2007, 71, 1476–1480 CrossRef CAS.
- X. F. Wang, Y. Zhou, J. J. Xu and H. Y. Chen, Adv. Funct. Mater., 2009, 19, 1444–1450 CrossRef CAS.
- D. J. Lin, J. Wu, F. Yan, S. Y. Deng and H. X. Ju, Anal. Chem., 2011, 83, 5214–5221 CrossRef CAS.
- X. Liu, L. Cheng, J. Lei and H. Ju, Analyst, 2008, 133, 1161–1163 RSC.
- X. Liu, L. Guo, L. Cheng and H. Ju, Talanta, 2009, 78, 691–694 CrossRef CAS.
- W. W. Yu, L. H. Qu, W. Z. Guo and X. G. Peng, Chem. Mater., 2003, 15, 2854–2860 CrossRef CAS.
- X. Liu, H. Jiang, J. P. Lei and H. X. Ju, Anal. Chem., 2007, 79, 8055–8060 CrossRef CAS.
- Y. Wang, J. Lu, L. H. Tang, H. X. Chang and J. H. Li, Anal. Chem., 2009, 81, 9710–9715 CrossRef CAS.
- L. H. Shen, X. X. Cui, H. L. Qi and C. X. Zhang, J. Phys. Chem. C, 2007, 111, 8172–8175 CAS.
- X. F. Hu, H. Y. Han, L. J. Hua and Z. H. Sheng, Biosens. Bioelectron., 2010, 25, 1843–1846 CrossRef CAS.
- J. Geng, B. Liu, G. F. Jie and J. J. Zhu, J. Nanosci. Nanotechnol., 2008, 9, 2387–2391 CrossRef.
- W. Wei, S. Y. Zhang, C. X. Fang, S. Q. Zhao, B. K. Jin, J. Y. Wua and Y. P. Tian, Solid State Sci., 2008, 10, 622–628 CrossRef CAS.
- X. F. Wang, J. J. Xu and H. Y. Chen, J. Phys. Chem. C, 2008, 112, 7151–7157 CAS.
- J. J. Miao, T. Ren, L. Dong, J. J. Zhu and H. Y. Chen, Small, 2005, 1, 802–805 CrossRef CAS.
- B. Zhou, B. Liu, L. P. Jiang and J. J. Zhu, Ultrason. Sonochem., 2007, 14, 229–234 CrossRef CAS.
- J. Geng, X. D. Jia and J. J. Zhu, CrystEngComm, 2011, 13, 193–198 RSC.
- Y. Shan, J. J. Xu and H. Y. Chen, Chem. Commun., 2010, 46, 4187–4189 RSC.
- Y. Shan, J. J. Xu and H. Y. Chen, Chem. Commun., 2009, 905–907 RSC.
- P. Wu, L. N. Miao, H. F. Wang, X. G. Shao and X. P. Yan, Angew. Chem., Int. Ed., 2011, 50, 8118–8121 CrossRef CAS.
- H. F. Wang, Y. Li, Y. Y. Wu, Y. He and X. P. Yan, Chem.–Eur. J., 2010, 16, 12988–12994 CrossRef CAS.
- P. Wu, L. N. Miao, H. F. Wang, X. G. Shao and X. P. Yan, Angew. Chem., Int. Ed., 2011, 50, 8118–8121 CrossRef CAS.
- Y. He, H. F. Wang and X. P. Yan, Anal. Chem., 2008, 80, 3832–3837 CrossRef CAS.
- H. F. Wang, Y. He, T. R. Ji and X. P. Yan, Anal. Chem., 2009, 81, 1615–1621 CrossRef CAS.
- P. Wu, Y. He, H. F. Wang and X. P. Yan, Anal. Chem., 2010, 82, 1427–1433 CrossRef CAS.
- L. Deng, Y. Shan, J. J. Xu and H. Y. Chen, Nanoscale, 2012, 4, 831–836 RSC.
- Y. Chen, Y. Y. Shen, D. Sun, H. Y. Zhang, D. B. Tian, J. R. Zhang and J. J. Zhu, Chem. Commun., 2011, 47, 11733–11735 RSC.
- J. P. Xie, Y. G. Zheng and J. Y. Ying, J. Am. Chem. Soc., 2009, 131, 888–889 CrossRef CAS.
- M. Yu, C. Zhou, J. Liu, J. D. Hankins and J. Zheng, J. Am. Chem. Soc., 2011, 133, 11014–11017 CrossRef CAS.
- J. Zhou, C. Booker, R. Li, X. Zhou, T. K. Sham, X. Sun and Z. Ding, J. Am. Chem. Soc., 2007, 129, 744–745 CrossRef CAS.
- C. M. Ritchie, K. R. Johnsen, J. R. Kiser, Y. Antoku, R. M. Dickson and J. T. Petty, J. Phys. Chem. C, 2007, 111, 175–181 CAS.
- P. R. O'Neill, L. R. Velazquez, D. G. Dunn, E. G. Gwinn and D. K. Fygenson, J. Phys. Chem. C, 2009, 113, 4229–4233 CAS.
- E. G. Gwinn, P. O'Neill, A. J. Guerrero, D. Bouwmeester and D. K. Fygenson, Adv. Mater., 2008, 20, 279–283 CrossRef CAS.
- Y. L. Liu, K. L. Ai, X. L. Cheng, L. H. Huo and L. H. Lu, Adv. Funct. Mater., 2010, 20, 951–956 CrossRef CAS.
- C. Y. Tian, J. J. Xu and H. Y. Chen, Chem. Commun., 2012, 48, 8234–8236 RSC.
- C. Y. Tian, W. W. Zhao, J. Wang, J. J. Xu and H. Y. Chen, Analyst, 2012, 137, 3070–3075 RSC.
- R. X. Zhang, L. Z. Fan, Y. P. Fang and S. H. Yang, J. Mater. Chem., 2008, 18, 4964–4970 RSC.
- J. Geng, B. Liu, L. Xu, F. N. Hu and J. J. Zhu, Langmuir, 2007, 23, 10286–10293 CrossRef CAS.
- C. J. Mao, D. C. Wang, H. C. Pan and J. J. Zhu, Ultrason. Sonochem., 2011, 18, 473–476 CrossRef CAS.
- M. Haase and H. Schafer, Angew. Chem., Int. Ed., 2011, 50, 5808–5829 CrossRef CAS.
- M. L. Yin, L. Wu, Z. H. Li, J. S. Ren and X. G. Qu, Nanoscale, 2012, 4, 400–404 RSC.
- Y. H. Wang, L. Bao, Z. H. Liu and D. W. Pang, Anal. Chem., 2011, 83, 8130–8137 CrossRef CAS.
- Q. Li, C. Zhang, J. Y. Zheng, Y. S. Zhao and J. Yao, Chem. Commun., 2012, 48, 85–87 RSC.
- A. Devadoss, L. Dennany, C. Dickinson, T. E. Keyes and R. J. Forster, Electrochem. Commun., 2012, 19, 43–45 CrossRef CAS.
- L. Dennany, P. C. Innis, G. G. Wallace and R. J. Forster, J. Phys. Chem. B, 2008, 112, 12907–12912 CrossRef CAS.
- L. Dennany, T. E. Keyes and R. J. Forster, Analyst, 2008, 133, 753–759 RSC.
- Y. L. Chang, R. E. Palacios, F. R. F. Fan, A. J. Bard and P. F. Barbara, J. Am. Chem. Soc., 2008, 130, 8906–8907 CrossRef CAS.
- S. Guo, O. Fabian, Y. L. Chang, J. T. Chen, W. M. Lackowski and P. F. Barbara, J. Am. Chem. Soc., 2011, 133, 11994–12000 CrossRef CAS.
- T. Ren, J. Z. Xu, Y. F. Tu, S. Xu and J. J. Zhu, Electrochem. Commun., 2005, 7, 5–9 CrossRef CAS.
- Z. H. Dai, J. Zhang, J. C. Bao, X. H. Huang and X. Y. Mo, J. Mater. Chem., 2007, 17, 1087–1093 RSC.
- Z. Fang, X. Lin, Y. F. Liu, Y. T. Fan, Y. G. Zhu, Y. H. Ni and X. W. Wei, J. Mater. Sci., 2010, 45, 6805–6811 CrossRef CAS.
- Y. Xu, C. Song, Y. Sun and D. Wang, Mater. Lett., 2011, 65, 1762–1764 CrossRef CAS.
- I. L. Medintz, M. H. Stewart, S. A. Trammell, K. Susumu, J. B. Delehanty, B. C. Mei, J. S. Melinger, J. B. Blanco-Canosa, P. E. Dawson and H. Mattoussi, Nat. Mater., 2010, 9, 676–684 CrossRef CAS.
- Q. Zhu, M. Han, H. S. Wang, L. L. Liu, J. C. Bao, Z. H. Dai and J. Shen, Analyst, 2010, 135, 2579–2584 RSC.
- M. Zhang, W. J. Dai, M. Yan, S. G. Ge, J. H. Yu, X. R. Song and W. Xu, Analyst, 2012, 137, 2112–2118 RSC.
- T. Wang, S. Y. Zhang, C. J. Mao, J. M. Song, H. L. Niu, B. K. Jin and Y. P. Tian, Biosens. Bioelectron., 2012, 31, 369–375 CrossRef CAS.
- S. N. Ding, J. J. Xu and H. Y. Chen, Chem. Commun., 2006, 3631–3633 RSC.
- Y. S. Guo, X. P. Jia and S. S. Zhang, Chem. Commun., 2011, 47, 725–727 RSC.
- L. H. Yang, J. Zhu, Y. Xu, W. Yun, R. Y. Zhang, P. G. He and Y. Z. Fang, Electroanalysis, 2011, 23, 1007–1012 CrossRef CAS.
- G. F. Jie, P. Liu, L. Wang and S. S. Zhang, Electrochem. Commun., 2010, 12, 22–26 CrossRef CAS.
- L. J. Hua, H. Y. Han and H. B. Chen, Electrochim. Acta, 2009, 54, 1389–1394 CrossRef CAS.
- Z. Guo, T. Hao, J. Duan, S. Wang and D. Wei, Talanta, 2012, 89, 27–32 CrossRef CAS.
- J. Wang, W. W. Zhao, X. R. Li, J. J. Xu and H. Y. Chen, Chem. Commun., 2012, 48, 6429–6431 RSC.
- J. Wang, H. Han, X. C. Jiang, L. Huang, L. N. Chen and N. Li, Anal. Chem., 2012, 84, 4893–4899 CrossRef CAS.
- G. Williams, B. Seger and P. V. Kamat, ACS Nano, 2008, 2, 1487–1491 CrossRef CAS.
- Y. T. Kim, J. H. Han, B. H. Hong and Y. U. Kwon, Adv. Mater., 2010, 22, 515–518 CrossRef CAS.
- D. A. Heller, H. Jin, B. M. Martinez, D. Patel, B. M. Miller, T. K. Yeung, P. V. Jena, C. Hobartner, T. Ha, S. K. Silverman and M. S. Strano, Nat. Nanotechnol., 2009, 4, 114–120 CrossRef CAS.
- C. H. Lu, H. H. Yang, C. L. Zhu, X. Chen and G. N. Chen, Angew. Chem., Int. Ed., 2009, 48, 4785–4787 CrossRef CAS.
- X. L. Dong, J. S. Cheng, J. H. Li and Y. S. Wang, Anal. Chem., 2010, 82, 6208–6214 CrossRef CAS.
- Y. Q. Wen, F. F. Xing, S. J. He, S. P. Song, L. L. Wang, Y. T. Long, D. Li and C. H. Fan, Chem. Commun., 2010, 46, 2596–2598 RSC.
- H. F. Dong, W. C. Gao, F. Yan, H. X. Ji and H. X. Ju, Anal. Chem., 2010, 82, 5511–5517 CrossRef CAS.
- M. Zhou, Y. L. Wang, Y. M. Zhai, J. F. Zhai, W. Ren, F. A. Wang and S. J. Dong, Chem.–Eur. J., 2009, 15, 6116–6120 CrossRef CAS.
- Z. J. Wang, X. Z. Zhou, J. Zhang, F. Boey and H. Zhang, J. Phys. Chem. C, 2009, 113, 14071–14075 CAS.
- H. L. Guo, X. F. Wang, Q. Y. Qian, F. B. Wang and X. H. Xia, ACS Nano, 2009, 3, 2653–2659 CrossRef CAS.
- C. Wang, E. Yifeng, L. Fan, S. Yang and Y. Li, J. Mater. Chem., 2009, 19, 3841–3846 RSC.
- Y. Shan, J. J. Xu and H. Y. Chen, Chem. Commun., 2010, 46, 5079–5081 RSC.
- J. Wang, Y. Shan, W. W. Zhao, J. J. Xu and H. Y. Chen, Anal. Chem., 2011, 83, 4004–4011 CrossRef CAS.
- H. Zhou, J. Liu, J. J. Xu and H. Y. Chen, Chem. Commun., 2011, 47, 8358–8360 RSC.
- F. Divsar and H. X. Ju, Chem. Commun., 2011, 47, 9879–9881 RSC.
- L. Dennany, M. Gerlach, S. O'Carroll, T. E. Keyes, R. J. Forster and P. Bertoncello, J. Mater. Chem., 2011, 21, 13984–13990 RSC.
- L. Hua, J. Zhou and H. Han, Electrochim. Acta, 2010, 55, 1265–1271 CrossRef CAS.
- Y. Li, M. Sun, F. Yang and X. Yang, Anal. Lett., 2010, 43, 2837–2847 CrossRef CAS.
- H. X. Ju, X. J. Zhang and J. Wang, NanoBiosensing–Principles, Development and Applications, Springer, NY, 2011 Search PubMed.
- M. Zhang, F. Wan, S. Wang, S. Ge, M. Yan and J. Yu, J. Lumin., 2012, 132, 938–943 CrossRef CAS.
- L. H. Zhang, L. Shang and S. J. Dong, Electrochem. Commun., 2008, 10, 1452–1454 CrossRef CAS.
- H. Han, Z. Sheng and J. Liang, Anal. Chim. Acta, 2007, 596, 73–78 CrossRef CAS.
- H. Jiang, H. P. Wang and M. X. Wang, Electrochim. Acta, 2010, 56, 553–558 CrossRef CAS.
- J. Wang, W. W. Zhao, C. Y. Tian, J. J. Xu and H. Y. Chen, Talanta, 2012, 89, 422–426 CrossRef CAS.
- C. G. Shi, J. J. Xu and H. Y. Chen, J. Electroanal. Chem., 2007, 610, 186–192 CrossRef CAS.
- M. Zhang, M. Yan, J. Yu, S. Ge, F. Wan and L. Ge, Anal. Methods, 2012, 4, 460–465 RSC.
- Y. Li, L. Liu, X. Fang, J. Bao, M. Han and H. Z. Dai, Electrochim. Acta, 2012, 65, 1–6 CrossRef CAS.
- L. F. Chen, Q. H. Cai, F. Luo, X. Chen, X. Zhu, B. Qiu, Z. Y. Lin and G. N. Chen, Chem. Commun., 2010, 46, 7751–7753 RSC.
- D. Y. Tian, C. F. Duan, W. Wang and H. Cui, Biosens. Bioelectron., 2010, 25, 2290–2295 CrossRef CAS.
- H. P. Huang and J. J. Zhu, Biosens. Bioelectron., 2009, 25, 927–930 CrossRef CAS.
- H. Huang, G. Jie, R. Cui and J.-J. Zhu, Electrochem. Commun., 2009, 11, 816–818 CrossRef CAS.
- X. Hu, R. Wang, Y. Ding, X. Zhang and W. Jin, Talanta, 2010, 80, 1737–1743 CrossRef CAS.
- H. P. Huang, Y. L. Tan, J. J. Shi, G. X. Liang and J. J. Zhu, Nanoscale, 2010, 2, 606–612 RSC.
- C. G. Shi, X. Shan, Z. Q. Pan, J. J. Xu, C. Lu, N. Bao and H. Y. Gu, Anal. Chem., 2012, 84, 3033–3038 CrossRef CAS.
- M. S. Wu, H. W. Shi, L. J. He, J. J. Xu and H. Y. Chen, Anal. Chem., 2012, 84, 4207–4213 CrossRef CAS.
- M. S. Wu, Q. G. Qian, J. J. Xu and H. Y. Chen, Anal. Chem., 2012, 84, 5407–5414 CrossRef CAS.
- S. N. Ding, J. J. Xu, D. Shan, B. H. Gao, H. X. Yang, Y. M. Sun and S. Cosnier, Electrochem. Commun., 2010, 12, 713–716 CrossRef CAS.
- J. L. Delaney, C. F. Hogan, J. Tian and W. Shen, Anal. Chem., 2011, 83, 1300–1306 CrossRef CAS.
- E. G. Hvastkovs, M. So, S. Krishnan, B. Bajrami, M. Tarun, I. Jansson, J. B. Schenkman and J. F. Rusling, Anal. Chem., 2007, 79, 1897–1906 CrossRef CAS.
- S. Krishnan, E. G. Hvastkovs, B. Bajrami, D. Choudhary, J. B. Schenkman and J. F. Rusling, Anal. Chem., 2008, 80, 5279–5285 CrossRef CAS.
- S. M. Pan, L. L. Zhao, J. B. Schenkman and J. F. Rusling, Anal. Chem., 2011, 83, 2754–2760 CrossRef CAS.
- N. P. Sardesai, J. C. Barron and J. F. Rusling, Anal. Chem., 2011, 83, 6698–6703 CrossRef CAS.
- S. Krishnan, E. G. Hvastkovs, B. Bajrami, I. Jansson, J. B. Schenkman and J. F. Rusling, Chem. Commun., 2007, 1713–1715 RSC.
- A. Venkatanarayanana, K. Crowleya, E. Lestini, T. E. Keyesa, J. F. Rusling and R. J. Forster, Biosens. Bioelectron., 2012, 31, 233–239 CrossRef.
- Z. J. Lin, X. M. Chen, T. T. Jia, X. D. Wang, Z. X. Xie, M. Oyama and X. Chen, Anal. Chem., 2009, 81, 830–833 CrossRef CAS.
- L. S. Dolci, S. Zanarini, L. D. Ciana, F. Paolucci and A. Roda, Anal. Chem., 2009, 81, 6234–6241 CrossRef CAS.
- M. Milutinovic, E. Suraniti, V. Studer, N. Mano, D. Manojlovic and N. Sojic, Chem. Commun., 2011, 47, 9125–9127 RSC.
- F. Deiss, C. N. LaFratta, M. Symer, T. M. Blicharz, N. Sojic and D. R. Walt, J. Am. Chem. Soc., 2009, 131, 6088–6089 CrossRef CAS.
- IUPAC Gold Book–electrogenerated chemiluminescence (ECL), http://goldbook.iupac.org/E01966.html.
- X. B. Yin, TrAC, Trends Anal. Chem., 2012, 33, 81–94 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |