Ultrasensitive detection of mRNA extracted from cancerous cells achieved by DNA rotaxane-based cross-rolling circle amplification†
Sai Bi*ab, Yangyang Cuib and Li Lib
aSchool of Chemistry and Chemical Engineering, Linyi University, Linyi 276005, P. R. China. E-mail: bisai11@126.com; Fax: +86 539 8766600; Tel: +86 539 8766600
bKey Laboratory of Biochemical Analysis, Ministry of Education, College of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Qingdao 266042, P. R. China
First published on 23rd October 2012
Abstract
An ultrasensitive and highly selective method for polymerase chain reaction-free (PCR-free) messenger RNA (mRNA) expression profiling is developed through a novel cross-rolling circle amplification (C-RCA) process based on DNA-rotaxane nanostructures. Two species of DNA pseudorotaxane (DPR) superstructures (DPR-I and DPR-II) are assembled by threading a linear DNA rod through a double-stranded DNA (dsDNA) ring containing two single-stranded gaps. In this assay, cDNA that is specific for β-actin (ACTB) mRNA is taken as a model analyte. Upon the introduction of the target cDNA, the cDNA and the biotin-modified primer are hybridized to the single-stranded regions of the DNA rod and the gap-ring, respectively. As a result, the DPR-I dethreads into free DNA macrocycle and a dumbbell-shaped DNA nanostructure. In the presence of DNA polymerase/dNTPs, two release-DNA on the DPR-I are replaced by polymerase with strand-displacement activity, which can act as the input of the DPR-II to trigger the dethreading of DPR-II and the RCA reaction, releasing another two specified release-DNA strands those in turn serve as the “mimic cDNA” for DPR-I. The C-RCA reaction then proceeds autonomously. To overcome the high background induced by hemin itself, the biotinylated rolling circle products are captured by streptavidin-coated MNPs, achieving a detection limit as low as 0.1 zmol cDNA. The assay also exhibits an excellent selectivity due to its unique DNA nanostructure fabricated through base pairing hybridization. The ACTB mRNA expression in mammary cancer cells (MCF-7) is successfully detected.
Introduction
Rotaxanes are a class of mechanically interlocked molecules consisting of a macrocycle that can thread readily over a dumbbell-shaped molecule, trapped kinetically by rigid and bulky stoppers.1 Since the mechanically interlocked components can move unhinderedly along the dumbbell-shaped molecules with large-amplitude motions relative to one another, a variety of rotaxanes have been made from a wide range of molecules and used in prototypical design features of components for nanoscale machinery.2–4 Recently, a novel kind of double-stranded DNA (dsDNA) pseudorotaxane is assembled, in which both the macrocycle and the dumbbell-shaped molecule are made of dsDNA, and the axle of the dumbbell is threaded through the macrocycle by base pairing hybridization.5 Upon the introduction of an oligonucleotide, the macrocycle is released from the axle, resulting in either the conversion of the pseudorotaxane into mechanically stable rotaxanes, or the disassembly of pseudorotaxane to a free macrocycle and a dumbbell. In comparison with the conventional rotaxanes, the dsDNA rotaxanes could certainly become attractive devices for fabricating more competent nanomachines due to their unique mechanical bonding motifs, opening a new horizon of DNA nanotechnology for biomolecular detection.Messenger RNA (mRNA) plays a significant role in numerous biologic processes, including the elongation of axonal growth cones, the guidance of fibroblast migration, and so on.6 For example, it has been found that aberrant mRNA processing can cause developmental and pathological abnormalities, such as viral infection, disrupted embryonic development, altered cell morphology, and susceptibility to cellular apoptosis.7 The alterations in mRNA expression can also be indicators of specific genetic diseases, especially cancer.8 Therefore, the sensitive and accurate detection of specific mRNA transcripts is of great significance to identify subtypes of a disease, which can be further used to determine the gene expression for early diagnosis and monitoring of treatment progress. So far, several techniques have been developed for mRNA expression profiling, such as Northern blot,9 ribonuclease protection analysis,10 microassay,11 electrochemical technique12 and fluorescent imaging.13,14 Recently, the most widely used method for the analysis of specific gene expression levels is the reverse transcription polymerase chain reaction (RT-PCR) assay through the synthesis of cDNA from a cellular mRNA.15
Rolling circle amplification (RCA) is an isothermal process, in which long single-stranded DNA (ssDNA) molecules with periodic sequence are synthesized by using a small circular ssDNA as template and a single DNA as primer for DNA polymerization reaction.16 By virtue of its unique advantages such as rapid analysis, high sensitivity and specificity, and significant amplification efficiency, RCA has become a novel amplification tool and has been widely applied in ultrasensitive detection of nucleic acid,17,18 proteins,19 viruses,20–22 and so on. Especially, the synthesis of horseradish peroxidase (HRP)-mimicking DNAzyme chains by RCA has become an attractive amplification strategy.20–22 DNAzymes have many advantages, such as catalytic activity, thermal stability, simple preparation and modification.23–25
Herein, an ultrasensitive and highly selective method for PCR-free mRNA expression profiling is developed through a novel cross-RCA (C-RCA) process based on DNA-rotaxane nanostructures. The interlocking approach is used to synthesize two DNA pseudorotaxane species (DPR-I and DPR-II) as prerequisites for the assembly of DNA-rotaxanes (DR). Upon the introduction of target cDNA that is specific for β-actin (ACTB) mRNA,26 it hybridizes the single-stranded region of DNA rod-I. Subsequently, primer-I binds to circle template-I to initiate the RCA-I process. During the RCA-I reaction, two release-DNA-I are displaced, which further act as targets to hybridize the single-stranded region of DNA rod-II to trigger the RCA-II process by employing circular DNA-II as template and biotinylated primer-II as primer. Similarly, during the RCA-II process, two release-DNA-II are released which in turn serve as the “mimic cDNA” for DPR-I and initiate another two RCA-I reactions. Then C-RCA is performed between DPR-I and DPR-II through the replacement of the corresponding release-DNA in the presence of biotinylated primers and DNA polymerase/dNTPs for the synthesis of hemin/G-quadruplex HRP-mimicking DNAzyme chains to catalyze the oxidation of 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS2−) by H2O2 to ABTS˙−. Additionally, the RCA products containing the complementary sequences to the single-stranded region of corresponding DNA rods can also initiate RCA reaction. Considering the high background induced by hemin itself in the ABTS2−/H2O2 UV-vis system, streptavidin-modified magnetic nanoparticles (MNPs) are employed to capture the rolling circle products (RCPs) via biotin–streptavidin specific recognition, achieving a superlow limit of detection of 0.1 zmol cDNA, which is comparable with that of PCR. As a potential analytical method that may facilitate clinical studies, the proposed assay is further applied to the detection of ACTB in mammary cancer cells (MCF-7).
It should be noted that streptavidin-modified MNPs used in this assay have many advantages. First, given the high background induced by hemin itself in the ABTS2−/H2O2 UV-vis system, streptavidin-coated MNPs are employed to capture the rolling circle products (RCPs) via biotin–streptavidin specific recognition, followed by incubating with hemin to generate series of DNAzymes. Thus, the excess hemin can be easily removed by magnetic separation, resulting in a low background signal. Second, the procedures of reaction and detection are separated by using MNPs, making mRNA analysis more accurate under optimum conditions. Moreover, as a magnetic carrier, MNPs may reduce the interference from the real samples which improves the anti-interference ability of the sensing system. Overall, MNPs used in this assay play a significant role in increasing the amplification efficiency.
As one of the most sensitive detection methods, PCR is no doubt a powerful technique for accurate and quantitative gene expression with the advantage of rapidity, high sensitivity and high reproducibility.27 In comparison with PCR, the proposed C-RCA method has the distinct advantages of its isothermal nature and high amplification efficiency, which can achieve high sensitivity that is comparable to PCR. Moreover, some isothermal methods have been developed so far, such as loop-mediated isothermal amplification (LAMP). LAMP amplifies DNA with high specificity and high efficiency, however they are often subject to the quantitative detection of LAMP products, which could effect the detection sensitivity.28 In this method, besides the advantages of isothermality and high amplification efficiency, the signal can be sensitively read out by UV-vis measurement through detecting the produced DNAzyme-catalyzed ABTS2−/H2O2 reaction, achieving a detection limit as low as 0.1 zmol cDNA.
Experimental section
Chemicals
The circular DNA and the other DNA oligonucleotides were synthesized by Takara Biotechnology Co., Ltd. (Dalian, China) and Sangon Biotechnology Co., Ltd. (Shanghai, China), respectively, and their sequences are listed in Table S1 (see ESI†). Klenow exo− DNA polymerase was provided by Fermentas (Canada). The deoxyribonucleoside 5′-triphosphates (dNTPs) were ordered from SBS Genetech Co., Ltd. (Beijing, China). H2O2 with analytical grade was obtained from Shanghai Chemical Reagent Company (Shanghai, China). Hemin, 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) disodium salt (ABTS2−) and 4-(2-hydroxyethyl)piperazine-1 ethanesulfonic acid sodium salt (HEPES) were purchased from Aladdin Chemistry Co. Ltd (China). Hemin was prepared as a stock solution (5.0 mM) with DMSO and stored in the dark at −20 °C. Dynabeads M-280 streptavidin-coated magnetic nanoparticles (MNPs) (10 mg mL−1) were ordered from Invitrogen Biotechnology Co. Ltd. (USA). Double-distilled, deionized water was used throughout the experiments. All chemicals were of analytical reagent grade and used without further purification.Cell lysis and RNA isolation
The MCF-7 cell lines were cultured according to the instructions of American Type Culture Collection in RPMI 1640 medium supplemented with 10% fetal bovine serum and 100 IU mL−1 penicillin–streptomycin, which were maintained at 37 °C in a 5% CO2-humidified air atmosphere. The cell density was counted with a hemocytometer. A ∼6 × 106 cells per mL suspension was centrifuged at 3500 rpm for 5 min in culture medium, washed once with PBS buffer, followed by spinning down at 3500 rpm for 5 min. The cell pellets were then suspended in 600 μL of lysis solution. Total RNA was prepared from MCF-7 cells using a High-purity Total RNA Rapid Extraction Kit (Shanghai Generay Biotech Co., Ltd., China) according to the manufacturer's instruction.Synthesis of cDNA from mRNA template
The mRNA was isolated from MCF-7 cells and then reverse transcribed to cDNA by using M-MuLV First Strand cDNA Synthesis Kit (Shanghai Sangon Biotechnology Co., Ltd., China) according to the manufacturer procedures. Briefly, mRNA samples with different amounts and DNA primers (20 pmol) were mixed and complemented up to 12 μL by RNase-free dd-H2O. After centrifugation, the mixture was heated on water-bath at 70 °C for 5 min, immediately followed by cooling down on an ice-bath for 10 s and centrifugation. The resulting mixture was placed on the ice-bath and 4 μL of 5× reaction buffer, 1 μL of RNase inhibitor (20 U μL−1) and 2 μL of dNTPs (10 mM) were successively added. Then the reaction mixture was incubated with gentle shaking at 37 °C for 5 min and 1 μL of M-MuLV reverse transcriptase (20 U μL−1) was added to make a final volume of 20 μL. The above mixture was reacted at 37 °C for 60 min, followed by incubating at 70 °C for 10 min to terminate the reaction. The resulting cDNA products were stored at −20 °C until further use.Fabrication of DNA-pseudorotaxane (DPR)
Taking the construction of DPR-I as example, the linear DNA rod-I (2.5 × 10−8 M), circle template-I (2.5 × 10−8 M), and release DNA-I (5.0 × 10−8) were mixed and annealed at 90 °C for 5 min, followed by incubating at 25 °C for 1 h, resulting in the fabrication of DPR-I. Similarly, DPR-II was constructed according to the above procedures.Detection of cDNA
A certain amount of cDNA was added to a mixture of DPR-I (5.0 × 10−9 M), DPR-II (5.0 × 10−9 M), biotinylated primer-I (5.0 × 10−9 M) and biotinylated primer-II (5.0 × 10−9 M) in 5 μL of RCA reaction buffer containing 0.4 U μL−1 Klenow exo− DNA polymerase and 1 mM dNTPs. The RCA reaction proceeded at 37 °C for 2 h, which was then cooled at 0 °C for 10 min to inactivate the polymerase. The biotinylated RCA products were reacted with 10 μL of streptavidin-modified MNPs at 37 °C for 30 min. After washing with TE buffer three times, the resulting MNPs were reacted with 4.0 × 10−7 M hemin solution to synthesize the DNAzyme chains. The excess hemin was readily removed by a magnetic field, followed by washing the MNPs with the same TE buffer for another three times and then dispersed in 850 μL of a buffer solution (25 mM HEPES, 20 mM KCl, 200 mM NaCl, Triton X-100 (0.05%, w/v) and DMSO (1%, v/v); pH 7.4). Subsequently, the resulting DNA–hemin complex on MNPs and 400 μL of ABTS2− (10 mM) were mixed in the same HEPES solution, followed by addition of 750 μL of H2O2 (5.3 mM). The UV-vis measurement was performed on a Cary50 UV-vis spectrophotometer (Varian, USA) after the addition of ABTS2− and H2O2 at 10 min in the wavelength range 200–800 nm.RT-PCR
RT-PCR was performed by the addition of 2 μL of synthesized cDNA, 2 μL of 10 × PCR buffer, 10 pmol upper primer, 10 pmol lower primer, 0.5 μL of dNTPs (10 mM each), 1.2 μL of Mg2+ (25 mM) and Taq DNA polymerase 1 U. The mixture was filled up to 20 μL by dd-H2O. After centrifugation, the mixture was heated to 94 °C for 5 min for the initial denaturation, followed by incubating in a MG25+ Thermocycler at 94 °C for 30 s, at 60 °C for 30 s, and at 72 °C for 30 s. The final extension was allowed to proceed for 5 min at 72 °C. The RT-PCR products were analyzed by nondenaturing polyacrylamide gel (15%) electrophoresis and ethidium bromide staining.Nondenaturing polyacrylamide gel electrophoresis (PAGE)
First, 15% polyacrylamide gel was prepared in Tris-acetate-EDTA (1× TAE) buffer (40 mM Tris, 20 mM acetic acid and 2 mM EDTA, pH 8.5). Subsequently, the samples were loaded in polyacrylamide gel with 6× loading buffer, followed by running at 110 V (constant voltage) for 1 h. Then the gels were stained with ethidium bromide (0.5 μg mL−1) in TAE for 30 min. Finally, images were obtained using a digital camera under UV illumination.Results and discussion
Principle of the assay
The principle of our DNA-rotaxane-based C-RCA strategy for colorimetric detection of mRNA is illustrated in Scheme 1. Structurally, the two kinds of DNA-pseudorotaxane (DPR) nanostructures consist of three parts. For example, DPR-I consists of circle template-I, release-DNA-I and linear DNA rod-I, while DPR-II consists of circle template-II, release-DNA-II and linear DNA rod-II. First, DPR-I and DPR-II are assembled by hybridizing complementary single-stranded regions of linear DNA rods to single-stranded gaps on dsDNA rings, respectively. In one constitutional unit, two hairpins on each end of the rod serve as mimic stoppers, resulting in a dumbbell-shaped DNA structure, which further fabricate the dsDNA-pseudorotaxane by threading the linear DNA rod through the corresponding gap-ring.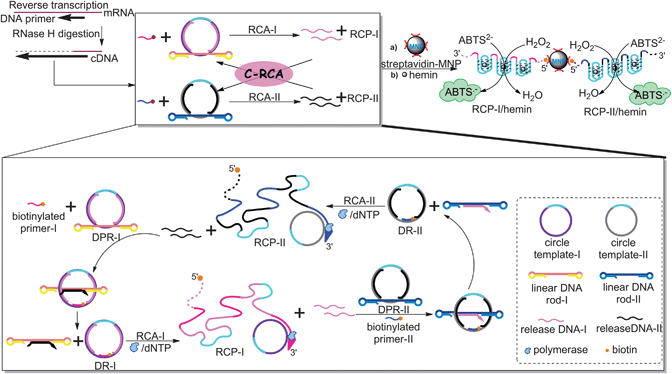 | ||
| Scheme 1 The C-RCA strategy for amplified assay of mRNA based on two kinds of DPR supermolecular structures with DNAzyme-based colorimetric detection through employing streptavidin–MNP to overcome the high signal background. | ||
Functionally, each part plays a significant role in the proposed amplification process. Upon the introduction of cDNA that is complementary to the single-stranded regions of the DNA rod-I, the DPR-I is converted into a “genuine” DNA rotaxane-I (DR-I) by hybridizing with the biotin-modified primer-I with 16-mer at the single-stranded region of the gap-ring. The resulting DR-I can move unhindered along the axle. According to previous study,5 the resulting rotaxane only exists as a short-lived intermediate, which rapidly disassembles into free DNA macrocycle and dumbbell-shaped DNA rod through dethreading. In addition, since the hairpins are not rigid enough to serve as efficient stoppers, the macrocycle can slip over the stopper hairpins, which further confirms that the DNA macrocycle has free translational mobility along the vector of the DNA axle. In this case, the RCA-I reaction is proceeded by employing circular DNA of the gap-ring as a template in the presence of DNA polymerase Klenow exo− and dNTPs. Here, the other single-stranded region of gap-ring is designed as the complementary sequence of DNAzyme. Thus, during the RCA reaction, the products of RCP-I containing thousands of repeated HRP-mimicking DNAzyme units are produced, which generate a catalytic signal upon complexing with hemin in the ABTS2−/H2O2 system. Meanwhile, in the process of primer extension, two specified release-DNA-I strands are displaced by polymerase with strand-displacement activity, which are complementary to the single-stranded regions of the DNA rod of DPR-II, and further trigger the RCA-II process by employing circular DNA of DPR-II as template and biotinylated primer-II as primer with the aid of DNA polymerase/dNTPs. Similarly, the RCA products RCP-II containing numerous DNAzymes are synthesized, while another two specified release-DNA-II strands are released, which in turn serve as the “mimic cDNA” for DPR-I and trigger yet another RCA-I. Subsequently, the reaction is autonomous and crossed to perform the cross-rolling circle amplification (C-RCA). After implementing the C-RCA reaction, the biotinylated RCPs are attached to the surface of streptavidin-coated MNPs via specific biotin–streptavidin interactions, followed by incubating with hemin to generate series of DNAzymes to catalyze the H2O2 oxidation of ABTS2− to the colored radical, ABTS˙− (λmax = 419 nm). In this way, the high background induced by free hemin could be easily eliminated through magnetic separation.
It should be noted that throughout the whole process, the RCA products – RCP-I and PCP-II containing the complementary sequences to single-stranded region of DNA rod-II and DNA rod-I, respectively – play key roles as triggers to form DR-II and DR-I, further initiating the RCA-II and RCA-I reactions, whose products can also undergo the proposed C-RCA (Scheme 2). This reaction pathway could make a great contribution to the improvement of the assay sensitivity.
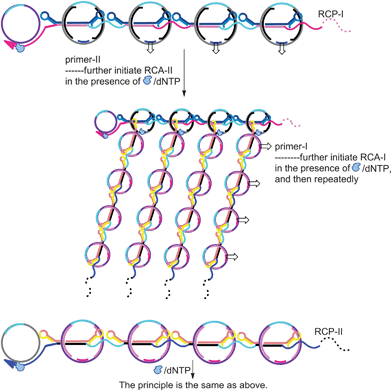 | ||
| Scheme 2 Schematic illustration of RCP-triggered transformation of DPRs to DRs through hybridizing the complementary sequences of RCP-I and RCP-II to the single-stranded region of DNA rods of DPR-II and DPR-I, while primers to the single-stranded gaps of respective DPRs, which further proceed the RCA reactions to enhance the amplification. | ||
In comparison with traditional RCA reaction, the major advantage of the proposed C-RCA is that the sensitivity could be significantly improved. For the traditional RCA, one target can only initiate one RCA process.17,20,21 However, in the proposed C-RCA, the target could be exponentially amplified by taking the displaced release-DNA and RCA products as “mimic targets” to further initiate RCA. Since the sensitivity of the assay is dependent on the amount of RCA products which assemble with hemin to generate DNAzymes to catalyze ABTS2−/H2O2, the substantially increased targets in C-RCA will lead to a significant amplification.
Polyacrylamide gel electrophoresis (PAGE)
The DPR supermolecular structures fabricated by threading linear DNA rods through corresponding gap-rings were characterized by nondenaturing polyacrylamide gel electrophoresis (PAGE). As shown in Fig. 1, lane 1, it is confirmed that the two kinds of DPRs (DPR-I and DPR-II) constructed could remain their stability under the experimental conditions. Upon the introduction of target cDNA accompanied by addition of primers, the DPR-I rapidly disassembled into free DNA macrocycle and linear DNA rods (Fig. 1, lane 2). Moreover, in the presence of Klenow exo− DNA polymerase/dNTPs, the C-RCA system started to operate until exhausting the total DPRs and primers (Fig. 1 lane 3), and the long strand products of the C-RCA reaction were too large to enter the nondenaturing gel by electrophoresis. The above results confirmed the feasibility of the proposed assay. | ||
| Fig. 1 Nondenaturing PAGE characterization. M: Marker. Lane 1: sample of DPR-I and DPR-II. Lane 2: as in lane 1, the DRP-I is disassembled into free DR-I and linear rod-I with subsequent addition of cDNA target and primers. Lane 3, the C-RCA results from the sample of lane 2 in the presence of Klenow exo− DNA polymerase/dNTPs. The DPRs completely disappear due to dethreading of the macrocycle. | ||
Sensitivity
The cDNA specific for β-actin was subsequently detected by the proposed C-RCA strategy. The signals were sensitively read out by UV-vis measurement to evaluate the sensitivity and linear range of the assay. According to the principle, the intensity of the detected colorimetric signal is correlated to the amounts of DNAzymes generated during the C-RCA, which are directly proportional to the amount of target cDNA. In order to make the reaction sufficient and ensure the sensitivity, excess DPRs and biotinylated primers (5.0 × 10−9 M for each) were added in the sensitivity experiment. In curve a of Fig. 2, a weak signal was observed in the absence of target (blank sample) even in the presence of primers and DNA polymerase/dNTPs, which further confirmed that the whole network was only triggered by target cDNA, followed by the C-RCA process and DNAzymes catalyzed colorimetric detection. As shown in Fig. 2, the UV-vis intensity increased with the increase of cDNA amount. A linear response can be obtained in a range of 0.1 zmol to 100 amol with a regression equation of ΔA = 0.0117log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) C + 0.2101 (ΔA is the relative UV-vis absorbance at a wavelength of 419 nm; C is the amount of cDNA, n = 7, R = 0.9984). The limit of detection (LOD) as low as 0.1 zmol was achieved. The precision of the assay was quantified by relative standard deviation (RSD) which was calculated to be less than 10% from 10 zmol cDNA (n = 11).
C + 0.2101 (ΔA is the relative UV-vis absorbance at a wavelength of 419 nm; C is the amount of cDNA, n = 7, R = 0.9984). The limit of detection (LOD) as low as 0.1 zmol was achieved. The precision of the assay was quantified by relative standard deviation (RSD) which was calculated to be less than 10% from 10 zmol cDNA (n = 11). | ||
| Fig. 2 (A) Colorimetric detection of cDNA for ACTB mRNA with different concentrations by the proposed C-RCA strategy. (B) The corresponding calibration curve of the relative UV-vis absorbance vs. the amount of cDNA. The relative UV-vis absorbance (ΔA) is calculated by A − A0, where A0 and A are the UV-vis absorbance without and with target cDNA, respectively. Error bars show standard deviations from the mean of three independent experiments. | ||
The ultrahigh sensitivity achieved in this study could mainly be attributed to the proposed C-RCA strategy. Although streptavidin–MNPs and biotin-primers used in this assay may increase the cost in the DNA amplification process, they can be easily produced and have been commercialized. More importantly, they play significant roles in overcoming high background and in amplifying the signal. Weighing the pros and cons, we use streptavidin-beads and biotin-primers to improve sensitivity. To evaluate the amplification efficiency of the C-RCA reaction, a control experiment was carried out by employing one circle template to perform RCA reaction. When the detection was performed by the single-stranded circle template, only a LOD of 10 zmol was obtained. Therefore, C-RCA played an essential role in enhancing the method sensitivity. Additionally, it should be noted that there is still a background in the blank by using MNPs, which could be attributed to an intrinsic enzyme mimetic activity of MNP that is similar to peroxidases.29 However, from the results, the signal peak of the blank could be decreased by employing streptavidin–MNPs to capture the biotinylated RCPs and magnetically remove the free hemin that was not intercalated with the synthesized DNAzymes, to reduce the high background, which makes a contribution to an improved sensitivity with a good reproducibility and validity. The LOD of 1.0 zmol cDNA under homogeneous reaction was ∼10-fold higher than that employing MNPs.
Specificity
To evaluate the method for discrimination of transcripts for mRNA expression profiling, we used the proposed C-RCA strategy and designed probe sequences for ACTB to analyze three other cancer-related transcripts, TERT, MYC (also known as cMyc) and ERBB2 (also known as HER2). The cDNA sequences for each transcript are listed in Table S1 (ESI†). As shown in Fig. 3, the colorimetric signal generated by 1.0 zmol cDNA for ACTB could obviously be distinguished from those produced by other transcripts, whereas no response over the control was detected when tested against other transcripts. So the proposed mRNA assay with a significant amplification exhibits an excellent specificity, probably due to the beneficial effect of hybridization selectivity for the transformation from DPR to DR. | ||
| Fig. 3 UV-vis signals generated by the proposed C-RCA strategy in the absence (blank sample) and presence of different ACTB, TERT, MYC and ERBB2 transcripts, respectively (1.0 zmol). | ||
Real sample assay
In human cancerous cells, mRNAs of interest are often present at relatively low copy number. Thus, high sensitivity and selectivity are essential to identify and quantify target mRNAs in cell homogenates. To veritably evaluate the assay, more complicated samples, such as cancer cells, needed to be analyzed to determine whether the assay could be useful to specifically and sensitively detect a unique mRNA in an actual sample. Here, we analyze the amount of ACTB in the total RNA of mammary cancer cells (MCF-7), which was prepared according to the previous report.26 cDNA that is created using DNA primer was synthesized from a cellular ACTB template using the M-MuLV First Strand cDNA Synthesis Kit, which could be measured without further purification even at a slightly reduced sensitivity. The high amplification efficiency and good response sensitivity of the proposed C-RCA strategy, as well as low background, enabled minimum detectable concentrations in the complex environment to be achieved without PCR. The concentration of ACTB in total RNA extracted from MCF-7 cells was calculated to be 117 ± 17 copies per cell, which were consistent with the previous studies of mRNA expression profiling,26 and were in good correlation with those of RT-PCR on the same sample (Table S2, ESI†). Given the greatly improved sensitivity, only a few cells are required to provide an adequate amount of mRNA, which can be easily detected by this method.So far, PCR and other DNA amplification methods have achieved single-cell analysis or the detection of a few copies of DNA.26,30,31 As an alternative method, the proposed assay has developed a new concept of C-RCA for gene detection based on DNA–rotaxane nanostructures. From the results of control experiments by employing one circle template to perform the traditional RCA and without using MNPs, the sensitivity of the assay is improved by 100-fold and 10-fold, respectively, which achieved a LOD as low as 0.1 zmol (∼60 copies) cDNA. Hence, the proposed C-RCA method is promising as an attractive alternative for gene expression profiling at single-cell level, opening the door to clinical diagnostics, particularly for early cancer diagnosis.
Conclusions
In summary, we have developed a C-RCA method for amplified assay of mRNA through the synthesis of numerous DNAzyme catalytic units based on DNA–rotaxane nanostructure. The association of the streptavidin–MNPs to capture biotinylated RCPs which could magnetically separate unreacted hemin, leads to a low background, achieving an ultrahigh sensitivity as low as 0.1 zmol cDNA specific for ACTB transcripts. Combining with the excellent selectivity owing to the specific base-pairing hybridization for the construction of DPR and transformation to DR, our amplification strategy is successfully applied to detect low levels of mRNA in cancerous cells without PCR. As an attractive alternative for mRNA expression profiling with high sensitivity, selectivity, and a wide dynamic range, the proposed assay opens up a new opportunity in the field of DNA nanotechnology-based bioanalysis, which also should be valuable to cancer diagLnostics and prognostics based on mRNA signatures or to discovery and functional study of new mRNA strands.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21105052), the University Doctoral New Teacher Foundation of the Ministry of Education (20113719120001), and the Foundation of State Key Laboratory of Electroanalytical Chemistry (SKLEAC201210).Notes and references
- C. A. Schalley, K. Beizai and F. Vögtle, Acc. Chem. Res., 2001, 34, 465–476 CrossRef CAS.
- F. Cacialli, J. S. Wilson, J. J. Michels, C. Daniel, C. Silva, R. H. Friend, N. Severin, P. Samorì, J. P. Rabe, M. J. O'Connell, P. N. Taylor and H. L. Anderson, Nat. Mater., 2002, 1, 160–164 CrossRef CAS.
- F. Aricó, J. D. Badjic, S. J. Cantrill, A. H. Flood, K. C.-F. Leung, Y. Liu and J. F. Stoddart, Top. Curr. Chem., 2005, 249, 203–259 Search PubMed.
- A. Fernandes, A. Viterisi, F. Coutrot, S. Potok, D. A. Leigh, V. Aucagne and S. Papot, Angew. Chem., Int. Ed., 2009, 48, 6443–6447 CrossRef CAS.
- D. Ackermann, T. L. Schmidt, J. S. Hannam, C. S. Purohit, A. Heckel and M. Famulok, Nat. Nanotechnol., 2010, 5, 436–442 CrossRef CAS.
- C. E. Holt and S. L. Bullock, Science, 2009, 326, 1212–1216 CrossRef CAS.
- A. Jayagopal, K. C. Halfpenny, J. W. Perez and D. W. Wright, J. Am. Chem. Soc., 2010, 132, 9789–9796 CrossRef CAS.
- S. R. Morris and L. A. Carey, Curr. Opin. Oncol., 2007, 19, 547–551 CrossRef CAS.
- F. Li, E. S. Barnathan and K. Karikó, Nucleic Acids Res., 1994, 22, 1764–1765 CrossRef CAS.
- A. J. Vidal-Puig, R. V. Considine, M. Jimenez-Liñan, A. Werman, W. J. Pories, J. F. Caro and J. S. Flier, J. Clin. Invest., 1997, 99, 2416–2422 CrossRef CAS.
- M. Taniguchi, K. Miura, H. Iwao and S. Yamanaka, Genomics, 2001, 71, 34–39 CrossRef CAS.
- J. Liu, H. Zhou, J.-J. Xu and H.-Y. Chen, Analyst, 2012, 137, 3940–3945 RSC.
- S. R. Harry, D. J. Hicks, K. I. Amiri and D. W. Wight, Chem. Commun., 2010, 46, 5557–5559 RSC.
- G. Qiao, L. Zhuo, Y. Gao, L. Yu, N. Li and B. Tang, Chem. Commun., 2011, 47, 7458–7460 RSC.
- A. Peixoto, M. Monteiro, B. Rocha and H. Veiga-Fernandes, Genome Res., 2004, 14, 1938–1947 CrossRef CAS.
- W. Zhao, M. M. Ali, M. A. Brook and Y. Li, Angew. Chem., Int. Ed., 2008, 47, 6330–6337 CrossRef CAS.
- Y. Cheng, X. Zhang, Z. Li, X. Jiao, Y. Wang and Y. Zhang, Angew. Chem., Int. Ed., 2009, 48, 3268–3272 CrossRef CAS.
- B. Zou, Y. Ma, H. Wu and G. Zhou, Analyst, 2012, 137, 729–734 RSC.
- W. Cheng, L. Ding, Y. Chen, F. Yan, H. Ju and Y. Yin, Chem. Commun., 2010, 46, 6720–6722 RSC.
- Y. Tian, Y. He and C. Mao, ChemBioChem, 2006, 7, 1862–1864 CrossRef CAS.
- Z. Cheglakov, Y. Weizmann, B. Basnar and I. Willner, Org. Biomol. Chem., 2007, 5, 223–225 CAS.
- D. M. Köster, D. Haselbach, H. Lehrach and H. Seitz, Mol. BioSyst., 2011, 7, 2882–2889 RSC.
- I. Willner, B. Shlyahovsky, M. Zayats and B. Willner, Chem. Soc. Rev., 2008, 37, 1153–1165 RSC.
- R. Fu, T. Li, S. S. Lee and H. G. Park, Anal. Chem., 2011, 83, 494–500 CrossRef CAS.
- K. He, W. Li, Z. Nie, Y. Huang, Z. Liu, L. Nie and S. Yao, Chem.–Eur. J., 2012, 18, 3992–3999 CrossRef CAS.
- C. Larsson, I. Grundberg, O. Söderberg and M. Nilsson, Nat. Methods, 2010, 7, 395–397 CrossRef CAS.
- P. Liu, X. Li, S. A. Greenspoon, J. R. Scherer and R. A. Mathies, Lab Chip, 2011, 11, 1041–1048 RSC.
- C. Li, Z. Li, H. Jia and J. Yan, Chem. Commun., 2011, 47, 2595–2597 RSC.
- Y. Du, B. Li, S. Guo, Z. Zhou, M. Zhou, E. Wang and S. Dong, Analyst, 2011, 136, 493–497 RSC.
- X. Chen, S. Roy, Y. Peng and Z. Gao, Anal. Chem., 2010, 82, 5958–5964 CrossRef CAS.
- H. M. T. Choi, J. Y. Chang, L. A. Trinh, J. E. Padilla, S. E. Fraser and N. A. Pierce, Nat. Biotechnol., 2010, 28, 1208–1212 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Oligonucleotide sequences used in the experiments, nondenaturing PAGE results of RT-PCR, and results of real sample assay. See DOI: 10.1039/c2an36118c |
| This journal is © The Royal Society of Chemistry 2013 |