A highly fluorescent pH sensing membrane for the alkaline pH range incorporating a BODIPY dye†
Mandy Hechta, Werner Krausb and Knut Rurack*a
aDiv. 1.9 Sensor Materials, BAM Federal Institute for Materials Research and Testing, Richard-Willstätter-Str. 11, D-12489 Berlin, Germany. E-mail: knut.rurack@bam.de; Fax: +49 30 8104 1157
bDiv. 1.3 Structural Analysis, BAM, Berlin, Germany
First published on 3rd October 2012
Abstract
A robust and re-usable dipstick-type fluorescent pH sensor for the alkaline pH range was developed by embedding a brightly fluorescent boron–dipyrromethene (BODIPY) dye bearing an acidic phenol moiety into a polyurethane matrix immobilized on a 3D epoxy-functionalized polymer support. The sensor strip has a dynamic working range of pH 10.0–13.1, i.e., operates in strongly basic media where pH glass electrodes can suffer from alkaline errors, and tolerates a high electrolyte background such as simulated seawater and sewage. This work describes the preparation of the sensing material and provides insight into the features that a hydrogel sensing membrane can bestow on an embedded pH-responsive dye by means of optical spectroscopic investigations.
Introduction
The pH glass electrode is by far the most commonly used pH sensor. A major limitation of glass electrodes however is their rather poor performance at the extremes of the pH scale.1 Regarding the strongly alkaline pH region, this is a significant disadvantage for applications for instance in the paper industry,2 nuclear fuel reprocessing,3,4 waste and waste water treatment,5–9 agriculture,10 leather processing,11,12 metal mining and finishing13–15 or microbial production processes involving alkaliophiles,16,17 all of them requiring the monitoring and/or adjustment of alkaline pH. Although new reports on pH sensors are continually published, most of them deal with biotechnological applications and thus concern pH sensors for the physiological range.18–22 Developments of pH sensors for extreme pH like pH > 10 are still rather rare.23–27 Fluorescence-based indicators offer several advantages over conventional electrochemical pH sensors, including sensitivity, proneness against alkaline errors, miniaturization, format adaptability and remote access. However, the development of optical sensors for the upper alkaline pH range of aqueous solutions remains challenging and their availability is still limited. 4,4-Difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY)28 dyes are a potent and versatile platform for the construction of pH-sensitive fluorescent probes.19,29,30 Moreover, BODIPYs have also proven to be suitable dyes for a number of other sensing applications,21,31–34 because of their outstanding characteristics such as excellent thermal and photochemical stability, relatively high molar absorption coefficients and high fluorescence quantum yields.35,36 In addition, the wide variety of synthetic pathways toward BODIPY dyes nowadays available renders the tailoring of desired spectroscopic responses and recognition chemistries facile. However, it is still difficult to obtain a BODIPY derivative which shows both good solubility and bright fluorescence in water. BODIPYs with hydroxyaryl subunits are promising tools for the measurement of pH in both chemical and biological systems.30 Transforming this suitability into an actual sensor format requires the immobilization of the pH-sensitive dye in a thin ion-permeable membrane. The latter however has to avoid detrimental effects such as self-quenching between dye molecules at high concentrations, insufficient permeability or mobility of the analyte in question and leaching of the indicator, in the case that steric embedding is preferred to covalent attachment because it can dispense with time-consuming pre-functionalization steps.In the present paper, we report on the synthesis and spectroscopic characterization of a pH-sensitive fluorescence sensor film for the basic pH range, in which common pH glass electrodes can show alkaline errors, based on the embedding of a BODIPY dye into a polyurethane hydrogel matrix. The polymer matrix creates an organic microenvironment comparable with an aqueous ethanolic solution, thus preserving the outstanding spectroscopic features of the BODIPY fluorophore.
Experimental section
Materials and methods
All reagents were obtained from commercial suppliers and were used without further purification unless otherwise indicated. All air- and moisture-sensitive reactions were carried out under argon atmosphere in oven-dried glassware. TLC was performed on Merck Silica Gel 60 F254 TLC plates with a fluorescent indicator at 254 nm excitation. Compounds were visualized under UV light of 254 nm. Column chromatography was carried out with Merck Silica gel 60 (0.040–0.063 mm) using dichloromethane (CH2Cl2) as an eluent. NMR spectra were recorded on a 400 MHz (100.6 MHz for 13C) Bruker AV 400 spectrometer or on a 300 MHz (75.6 MHZ for 13C) Bruker DPX 300 spectrometer at 27 °C using residual protonated solvent signals as internal standard (1H: δ(CDCl3) = 7.26 ppm and 13C: δ(CDCl3) = 77.16 ppm). Assignments are based on chemical shifts and/or DEPT spectra (Ar is used as abbreviation for assigning aromatic moieties). Mass spectra were measured on a Waters LCT Premier XE. UPLC was performed with a Waters UPLC Acquity equipped with a Waters LCT Premier XE mass detector for UPLC-MS, with Waters Alliance systems (consisting of a Waters Separations Module 2695, a Waters Diode Array detector 996 and a Waters Mass Detector ZQ 2000) equipped with a Acquity BEH C18 (2.1 × 50 mm) column, and with Shimadzu LC-10A systems equipped with a photodiode array detector (PAD). UV-vis absorption spectra were recorded on a Specord 210-Plus spectrophotometer from Analytik Jena AG. Steady-state fluorescence measurements were carried out on a FluoroMax-4 spectrofluorometer from Horiba Jobin-Yvon Inc., New Jersey, using standard 10 mm path length quartz or PMMA cells. Fluorescence lifetimes were determined with a unique customized laser impulse fluorometer with picosecond time resolution described elsewhere.37,38 The fluorescence lifetime profiles were analyzed with a PC using the software package FLA900 (Edinburgh Instruments).37,38 Environmental scanning electron microscope (ESEM) measurements were performed with a XL 30 ESEM from FEI. All samples were coated with Aurum (Scancoat Six Sputter Coater from Edwards) by sputter coating for 90 s with a deposition rate of 15 nm min−1.All the solvents employed for the spectroscopic measurements were of UV spectroscopic grade (Aldrich). Schott Nexterion Slide E and PolyAn 3D-Epoxy Slides were used as solid supports for the dye-doped hydrogel and were cut into 10 mm strips which fit into commercially available PMMA cells. The hydrogel HydroMed D4 (ether-based hydrophilic urethane, water content 50%) was received from AdvanSource Biomaterials Corporation, Wilmington, MA.
Preparation of 1
3.0 mmol of 4-hydroxybenzaldehyde (0.37 g, 1.0 eq.) and 6.6 mmol of 3-ethyl-2,4-dimethylpyrrole (0.813 g, 2.2 eq.) were dissolved in 90 mL of absolute CH2Cl2 under argon atmosphere. 10 drops of trifluoroacetic acid were added and the solution was stirred at room temperature until 4-hydroxybenzaldehyde was completely consumed, which was monitored by TLC. Then, a solution of 3.0 mmol 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (0.68 g, 1.0 eq.) in 120 mL CH2Cl2 was added. After stirring for another 4 h, 18 mL of triethylamine (0.13 mol) and 18 mL of BF3·OEt2 (0.15 mol) were added dropwise into the mixture while cooling in an ice-bath. The reaction mixture was kept for stirring at RT overnight and then filtered through a celite pad. The residue was washed with 50 mL of CH2Cl2, evaporated to dryness and re-dissolved in 100 mL of CH2Cl2. The resulting solution was washed with 100 mL of aq. 5% NaHCO3 solution (w/w) and water (2 × 100 mL). The combined aq. phases were washed with CH2Cl2 again until the resulting organic solution was only slightly orange-coloured. The separated organic phases were dried over Na2SO4 and concentrated under vacuum. The crude product was purified by column chromatography on silica using CH2Cl2 as an eluent to give compound 1 as shiny, reddish-orange crystals (536 mg, 45%).1H NMR (CDCl3): δ = 0.98 (t, 6H, J = 7.6, 2 × CH2–CH3), 1.35 (s, 6H, C–CH3), 2.30 (q, 4H, J = 7.6, 2 × CH2–CH3), 2.53 (s, 6H, 2 × N–C–CH3), 6.94 (m, 2H, CHar), 7.12 (m, 2H, CHar). 13C NMR (CDCl3): δ= 0.98 (2 × CH2–CH3), 1.35 (C–CH3), 2.30 (2 × CH2–CH3), 2.53 (2 × N–C–CH3), 6.94 (CHar), 7.12 (CHar); for spectra, see Fig. S1, ESI.† MS: m/z calcd: C23H27BF2N2O [M]+: 396.2185, found: 396.2248.
2 was taken from a batch prepared earlier by us;39 since 2 is one of our internal reference dyes it is regularly checked for stability and purity.
Molar absorption coefficient and fluorescence quantum yield
The molar absorption coefficient ε(λex) was determined from three separately weighed and dissolved stock solutions in a repeat determination of each sample (N = 6 independent measurements). The dilution was chosen in such a way that the absorbance of the sample solutions equalled 0.5. The fluorescence quantum yields (Φf) were determined relative to dye 2 in MeCN (Φf = 0.87).39 The fluorescence spectra presented here were spectrally corrected.X-ray analysis
Single crystals of compound 1 suitable for X-ray diffraction analysis were obtained by slow crystallization in MeOH/CH2Cl2.Single crystal X-ray data collection was carried out on a Bruker AXS SMART diffractometer at room temperature using Mo Kα radiation (λ = 0.71073 Å), monochromatised by a graphite crystal. The data reduction was performed by using the Bruker AXS SAINT and SADABS packages. The structure was solved by direct methods and refined by full-matrix least squares calculation using SHELX.40,41 Anisotropic thermal parameters were employed for non-hydrogen atoms. The hydrogen atoms were treated isotropically with Uiso = 1.2 times the Ueq. value of the parent atom. The hydrogen of the OH-group was refined with the help of the rest electron density. Crystal data and refinement details are summarized in Tables S1 and S2, ESI.†
Fabrication of membrane-coated strips
The dye-doped hydrogel was prepared from 1.0 g HydroMed D4 dissolved in a mixture of 10.8 g EtOH and 1.2 g H2O. Starting from a stock solution of 1 in CH2Cl2 (c = 2.51 mM), 796.8 μL of the stock solution were added to the viscous hydrogel solution to achieve a dye concentration of 2 μmol g−1 of polymer.42 The mixture was vigorously stirred at room temperature for 1 h. The polymer strips were dip-coated into the hydrogel solution and air-dried for 3 h prior to use. To reduce the film thickness or to obtain reduced dye concentrations in the film, hydrogels were prepared as described but with the compositions listed in Table 1.X-ray photoelectron spectroscopy
XPS measurements were carried out with an AXIS Ultra DLD photoelectron spectrometer manufactured by Kratos Analytical (Manchester, UK). XPS spectra were recorded using monochromated Al Kα excitation at pass energies of 80 eV for survey and 20 eV for high-resolution core-level spectra. Additionally the charge neutralization system was applied. The electron emission angle was 0° and the source-to-analyzer angle was 60°. The binding energy scale of the instrument was calibrated following a Kratos Analytical procedure which uses ISO 15472 binding energy (BE) data.43,44 The binding energy scale was corrected for static charging by using BE = 285.0 eV for the aliphatic C 1s component.45 High resolution C 1s core level spectra were analyzed using the CasaXPS peak fit program (Casa Software Ltd., Teignmouth). In curve fitting of C 1s spectra, full widths at half maximum were constrained to be equal resulting in values ranging from 1.2–1.4 eV. A Gaussian/Lorentzian product function peak shape model (G/L = 30) was used in combination with a Shirley background.Measurements of pH
For every step of the pH titration of neat 1 in solution, small amounts of aq. KOH (0.02–2 M) were added (micro-litre pipette, Eppendorf) to a solution (2.5 mL) containing the dye (1.39 × 10−7 M) in a mixed solvent of H2O and EtOH 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v). The mixture was stirred and the pH value was monitored with a digital pH meter (pH lab 827, Metrohm GmbH) equipped with a glass electrode (Biotrode). For comparison, pH measurements were also performed with a second digital pH meter (WTW pH 537), equipped with a glass electrode InLab 423 (Mettler Toledo). Calibration of the instruments was performed with standard aqueous solutions of pH 4.00, 7.00 and 9.00 from Metrohm GmbH. The measurement uncertainties of the pH-electrodes amount to ≤±0.03 pH. For pH measurements with the sensor, the membrane-coated strip was placed in a cuvette filled with water and small amounts of an aq. KOH (0.1 M) solution were added while stirring for every step of the pH titration. The final concentration of KOH in the aqueous solution was 0.01 M.
:1 (v/v). The mixture was stirred and the pH value was monitored with a digital pH meter (pH lab 827, Metrohm GmbH) equipped with a glass electrode (Biotrode). For comparison, pH measurements were also performed with a second digital pH meter (WTW pH 537), equipped with a glass electrode InLab 423 (Mettler Toledo). Calibration of the instruments was performed with standard aqueous solutions of pH 4.00, 7.00 and 9.00 from Metrohm GmbH. The measurement uncertainties of the pH-electrodes amount to ≤±0.03 pH. For pH measurements with the sensor, the membrane-coated strip was placed in a cuvette filled with water and small amounts of an aq. KOH (0.1 M) solution were added while stirring for every step of the pH titration. The final concentration of KOH in the aqueous solution was 0.01 M.Interference studies
Various anions (H2PO4−, HSO4−, I−, Cl−, Br−, F−, NO3−, SCN−, benzoate as their tetrabutylammonium salts dissolved in MeCN or in EtOH–H2O (1:1)) were added to solutions of 1 (ca. 1 × 10−6 M) at a 5-fold molar excess while monitoring the spectroscopic changes in absorption and fluorescence. Similar experiments with the strip were carried out in analogy. Simulated seawater compositions prepared from commercially available marine salt (concentration of 1–3% per weight) were used for the studies concerning electrolyte background.Reversibility
The reversibility of the response of the sensor film was examined using an aq. KOH solution (0.01 M, pH 12.14) and water of pH 6.43. The fluorescence intensity of the film was measured in H2O first, then the sensor was dipped into the basic solution at room temperature for 5 s, and the emission of the film was measured again. The film was washed with water and air dried. Finally, the emission of the film was re-measured in H2O. The above processes were repeated 5 times.Cell phone test
The substrate was prepared by coating half of the strip with a hydrogel doped with 1 and the other half of the substrate with a hydrogel containing dye 2.39 BODIPY 2 serves as a reference dye, possessing the same BODIPY core and the meso-phenyl ring as 1 yet lacking a pH-sensitive functional group. The substrate was immersed in aq. KOH solutions of pH = 9.0, 10.5, 11.2 and 13.0, respectively, and photographs were taken with a cell phone with LED-flash and a 5 megapixel camera.Results and discussion
Synthesis and crystal structure of 1
The BODIPY derivative 1 was synthesized in 45% yield by condensation of 4-hydroxybenzaldehyde with two equivalents of kryptopyrrole using TFA as a catalyst, followed by oxidation with DDQ, deprotonation with TEA and finally complexation with boron trifluoride at room temperature according to literature reports (Fig. 1).30,46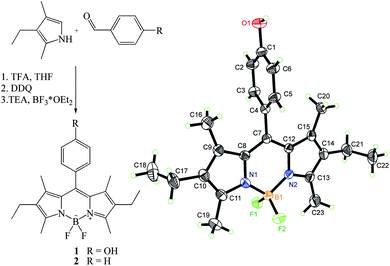 | ||
| Fig. 1 Left: synthesis of 1; right: crystal structure of 1 as derived from the X-ray analysis (thermal ellipsoid plots are drawn at the 30% probability level). | ||
X-ray analysis of 1 revealed that the dye crystallizes in the monoclinic space group P21/n with one molecule in the asymmetric unit and four molecules in the unit cell. The three-dimensional structure of 1 is illustrated in Fig. 1, together with the atomic labelling scheme. The X-ray data show that the meso-phenyl ring is virtually perpendicular to the plane of the BODIPY core with a dihedral angle of 87.02(6)° and that the central six-membered ring of the core is almost coplanar with the adjacent five-membered rings with the average deviation from the mean plane being 0.0031(2) Å. The same accounts for the two pyrrole rings, the four methyl groups and the two ethylene carbons, with a maximum deviation of 0.081(2) Å from the least-squares mean plane for 18 atoms. These findings are in line with previous reports on highly internally twisted 1,7-alkyl-substituted BODIPYs.47,48
Spectroscopic properties of 1
Table 2 shows the spectroscopic features of 1 investigated by absorption as well as steady-state and time-resolved fluorescence spectroscopy in various solvents with increasing polarity from hexane to water. The maxima of the absorption and fluorescence spectra are virtually independent of solvent polarity except for water in which a broadening of the absorption band is observed (Fig. 2). This effect can be attributed to the moderate solubility of the hydrophobic dye in water, accompanied by the tendency for dimer formation, with microsolvation effects being possibly also involved. The weak solvatochromism in organic solvents observed for 1 is advantageous because it reveals that the dye is insensitive to a change in an organic environment49 and suggests that electrolyte effects should also not play a prominent role. Notably, in EtOH–H2O we found 1 to be extremely brightly fluorescent in comparison to neat organic solvents. The molar absorption coefficient of 71900 ± 630 M−1 cm−1 in acetonitrile is characteristically high. The fluorescence decay curves could be described by a single exponential fit (χ2 < 1.2) and the fluorescence decay times (τf) lie between 4.3 and 4.7 ns in organic solvents, and 5.6 ns in the ethanolic aqueous solution. The rate constants for radiative (kr) and non-radiative (knr) deactivation were calculated using the standard expressions shown in Table 2. In water, the fluorescence is distinctly quenched and the lifetime could only be fitted to a non-exponential decay. In combination with the above mentioned spectral behaviour in water, such a quenched fluorescence also supports dimer formation, the strength of the fluorescence of the dimer species being still a matter of debate.50,51 The complicated excited-state behaviour of 1 in water disfavours a straightforward use of 1 as a pH indicator, because monomer–dimer equilibria tend to depend strongly on the electrolyte content, pH and other environmental features. In addition, the quenched fluorescence is unfavourable with regard to analytical performance.
| Solventa | λabs/nm | ελabs/M−1 cm−1 | λem/nm | Φf | τf/ns | krb/108 s−1 | knrb/108 s−1 |
|---|---|---|---|---|---|---|---|
| a Hex = n-hexane, DEE = diethyl ether, THF = tetrahydrofuran.b kr = Φf × τf−1, knr = (1 − Φf) × τf−1; measurement uncertainties amount to ±0.01 × 108 s−1.c Not determined.d Global fluorescence quantum yield of the species mixture in water when excited at 495 nm.e The longest lifetime component of multi-exponential fit, attributed to the monomeric form of 1.f Not calculated because of species diversity. Further investigations are under way to shed more light on this behaviour. | |||||||
| Hex | 524 | n.d.c | 531 | 0.82 | 4.29 | 1.9 | 0.4 |
| DEE | 521 | n.d. | 529 | 0.91 | 4.72 | 1.9 | 0.2 |
| THF | 523 | n.d. | 531 | 0.92 | 4.57 | 2.0 | 0.2 |
| MeCN | 521 | 71900 | 531 | 0.83 | 4.72 | 1.8 | 0.4 |
| H2O/EtOH | 523 | n.d. | 532 | 0.97 | 5.57 | 1.7 | 0.1 |
| H2O | 523 | n.d. | 532 | 0.12d | 2.67e | f | f |
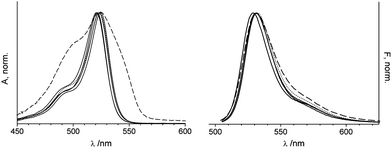 | ||
| Fig. 2 Normalized absorption (left) and fluorescence spectra (right) of 1 in different solvents (excitation at 495 nm). Hex, DEE, THF and MeCN as solid lines, H2O–EtOH as dotted lines and H2O as dashed lines. | ||
pH-dependent absorption and fluorescence spectroscopy of 1
As mentioned above, the meso-phenol moiety of 1 is sensitive to the environmental pH and can be deprotonated to the phenolate anion in basic media. Because highly electron-rich meso-substituents commonly quench the fluorescence of the BODIPY core through an efficient electron transfer process,46,52 an increase in pH should gradually switch off the fluorescence of 1. As is depicted in Fig. 3, almost a complete deprotonation of the phenolic OH group occurs above pH 13 indicated by the low fluorescence intensity. The effect of pH was assessed by altering the solution pH from 7 to 13 through successive addition of aq. KOH in ethanol–water 1:1 to avoid the formation of aggregates. The spectrophotometric and fluorometric pH titration spectra in Fig. 3 reveal that the wavelength of the emission maximum remains unchanged, despite the strong signal decrease; the fluorescence quantum yield of 1− lies below 10−4. In addition, the absorption maximum was shifted to shorter wavelengths, in agreement with the influence that a change in the inductive effect exerts on the HOMO/LUMO of the BODIPY core when converting a phenol into a phenolate moiety (see the Theoretical section in the ESI†). The protonation constant was determined to be pKa 9.98 ± 0.02 by Boltzmann curve-fitting as a function of pH using the fluorescence intensity at 535 nm. The dynamic sensing range spans 3 pH units, 8.75–11.65. It has to be mentioned that for an accurate determination of the pKa a correction by −0.2 according to ref. 53 is necessary to take into account the differences in proton activity coefficients and liquid junction potentials between the ethanol–water solvent mixture of the sample and the aqueous calibration solution.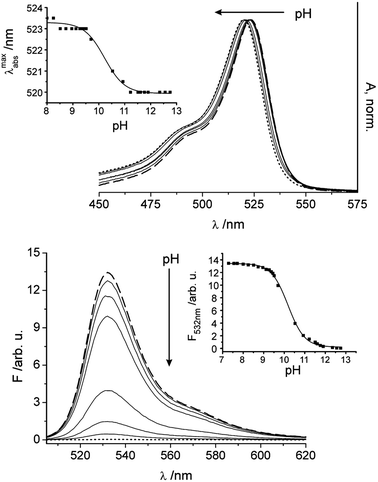 | ||
| Fig. 3 Dependence of the absorption and fluorescence of 1 on a change in pH from 7.3–12.8 upon addition of aq. KOH solutions (λex = 495 nm, c1 = 1.39 × 10−6 M, EtOH–H2O); the spectrum at pH 7.3 corresponds to the dashed line, the spectrum at pH 12.8 to the dotted line; and selected intermediate steps shown as solid lines. Insets show the corresponding titration curves when plotting the shift of the absorption maximum (top) and the fluorescence intensity at the emission maximum (bottom) as a function of pH. | ||
Synthesis and properties of a sensory film containing 1
The fluorescent thin film was prepared by simple dip coating of tailored 3D epoxy-functionalized polymer slides into the hydrogel–dye solution. After drying, the sensor was very robust and easy to apply. The thickness of the sensor layer was determined to be 9.3 ± 0.4 μm by ESEM through coating of similarly functionalized glass slides instead of polymer supports (Fig. 4). The replacement of the polymer support with glass was necessary due to the lack of contrast in the case of a hydrogel polymer on a polymer support (Fig. S2d, ESI†). | ||
| Fig. 4 ESEM image of a hydrogel film on a glass support and determination of the layer thickness. | ||
An effective dye concentration of 0.81 mM in the dried hydrogel layer was calculated from the molar absorption coefficient and the thickness of the layer.
A reduction of the film thickness down to 4 μm can be reproducibly realized by adjusting the initial amount of hydrogel (Fig. S2b and c†). As expected, the fluorescence signal decreases with the decrease in film thickness when the concentration of the dye is kept constant (Fig. S3†). In contrast to the applied 3D epoxypolymer substrate, plain and epoxysilane-functionalized glass surfaces showed no stable conjugation to the hydrogel. Films on these substrates were detached upon exposure to aqueous sample solutions (Fig. S2a†). Additional XPS investigations showed that the polymer support expresses a much higher number of C–O units at the surface than the glass slide, presumably influencing the adhesive forces between the support and the sensor layer (Table S1†).
Regarding the spectroscopic behaviour of 1 as shown in Fig. 1 and Table 2, the absorption maximum of the sensor material is centred at 525 nm in water and the emission at 534 nm, comparable to that of 1 in an organic microenvironment or in EtOH–H2O. In addition, the fluorescence lifetime of 1 amounts to 5.56 ns in the hydrogel film, similar to that of 1 in an ethanol–water mixture. Because the fluorescence decay was found to be mono-exponential, 1 seems to be distributed in a homogeneous environment in the film. The hydrogel matrix thus leads to a beneficial effect and preserves the exceptional features of 1 in its protonated state.
An investigation of the robustness and precision between batch preparations is included below in the Surface water and sewage section.
Performance of the sensor
The potential of the supported fluorescent polyurethane hydrogel matrix incorporating 1 for application purposes was assessed by monitoring the membrane's response toward various pH under different environmental settings. When the film-coated dipsticks are titrated with an aqueous potassium hydroxide solution in pure water, absorption and fluorescence changes like those shown in Fig. 5 are observed. As expected, the fluorescence intensity decreases as a function of pH, with similar slight or absent shifts of the absorption and emission maxima, constituting another advantageous feature of the composite membrane. The pH titration curve of the sensor exhibits the typical sigmoidal shape and the pKa was determined to be 11.44 ± 0.01. Presumably attributed to the interaction of the polyurethane matrix with the dye, the response of the sensor material lies at even more alkaline pH, rendering the material very suitable for this extreme pH range. In addition, a dynamic working range of 10.02–13.12 could be established. Another important feature of the physical entrapment of the hydrophobic dye in the hydrogel matrix is that the shortcomings of aggregation and low solubility in aqueous environments can be nicely overcome. The absence of any fluorescence ascribable to 1 from a solution in which the sensor was incubated for 300 min showed that the film does not suffer from leaching of the dye due to swelling of the hydrogel. Regarding comparability of the fluorometrically obtained results with pH data recorded with the two digital pH meters employed, the sensor strips showed excellent agreement of all three devices yielding matching pH within ±0.05 pH units.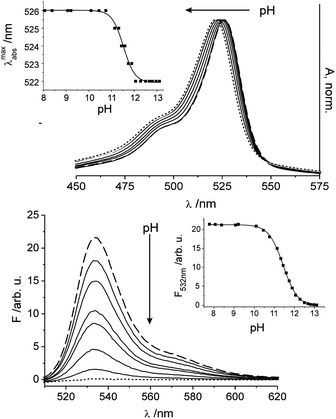 | ||
| Fig. 5 Dependence of the absorption (top) and fluorescence (bottom) spectra of a sensor film containing 1 in water on changes in pH under alkaline conditions upon addition of small amounts of an aq. KOH solution (0.1–2.0 M) with excitation at 495 nm; the spectrum at pH 8.2 corresponds to the dashed line, the spectrum at pH 13.2 to the dotted line; and selected intermediate steps shown as solid lines. Insets show the corresponding titration curves when plotting the shift of the absorption maximum (top) and the fluorescence intensity at the emission maximum (bottom) as a function of pH. | ||
Effect of anions and electrolyte background
It is known from the literature52 that basic inorganic and organic anions can lead to a deprotonation of an organic dye that carries a potentially acidic group in organic solvents in which the anions are much less solvated than in water. Such a behaviour can potentially interfere with the desired pH indication reaction. Since the spectroscopic properties of 1 in the hydrogel matrix are more reminiscent of a (mixed) organic than a neat aqueous environment, we studied the influence of potential interferents for neat and embedded 1. In organic solvents such as acetonitrile, 1 indeed suffers from deprotonation when basic anions such as fluoride, dihydrogen phosphate or benzoate are present at a 5-fold molar excess, discernible from the quenching of fluorescence shown in Fig. 6. In contrast, already in EtOH–H2O no remarkable changes in fluorescence intensity could be detected suggesting that the anions are well solvated (see Fig. S4 and S5, ESI† for the corresponding absorption spectra).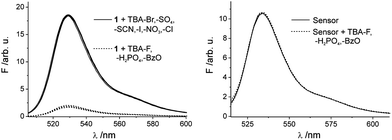 | ||
| Fig. 6 Fluorescence spectra of 1 in MeCN (left) upon addition of various anions and fluorescence spectra of the sensor film in water (right) upon addition of relevant anions H2PO4−, F− and BzO− (canion = 7.5 × 10−6 M). | ||
When stepping to the sensory film and monitoring its fluorescence in water in the presence of the most potent interferents in organic solution, Fig. 6 reveals that these species are spectroscopically silent under realistic sensing conditions, despite the different microenvironments in the hydrogel compared with water. Furthermore, we could demonstrate that the performance of the sensor film is unaffected by other ions using a simulated seawater composition of 3% (w/w) which corresponds to a saline concentration in the Atlantic.
Reversibility
To test the reversibility of the sensor film and to show the robustness against highly basic conditions the film was dipped alternately into an alkaline solution (pH = 12.14), which results in deprotonation of the dye associated with quenching of the fluorescence, and water of pH = 6.43, whereby the fluorescence intensity should return to its initial value at the end of each cycle (Fig. 7). Switching between the fluorescent on/off states can thus be repeated without decomposition of the membrane or the embedded dye.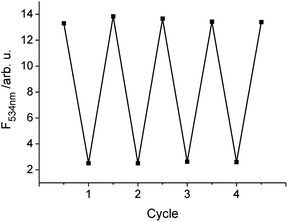 | ||
| Fig. 7 Reversibility of the fluorescence response of the sensor film upon successive deprotonation/protonation by dipping it into an alkaline solution of pH 12.14 and water of pH 6.43. | ||
Surface water and sewage
To evaluate the performance of the strips under realistic conditions and to assess the robustness and precision between batch preparations, three strips from individual batches were tested on three real samples, surface water from an inner city canal (sample 1) as well as influent (sample 2) and effluent (sample 3) water from a sewage treatment plant. The reproducibility in the fluorescence readings between the strips was good with a relative uncertainty of ≤2.2% (Table S2†), no matter if the pH of the samples was close to neutral or basic after spiking with 0.1 M KOH. A representative selection of fluorescence spectra is shown in Fig. 8. Since the initial pH of sample 2 was slightly higher than that of samples 1 and 3, the spiking with the same amount of alkaline solution led to a slightly stronger quenching in this case. For all cases, the optical data were again in good agreement (±0.05 pH units) with the electrochemical data.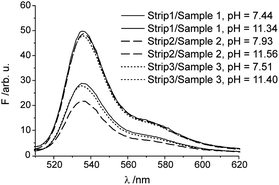 | ||
| Fig. 8 Representative fluorescence spectra of selected combinations of test strips and samples; the determined pH is indicated. For additional details, see text and Table S2.† | ||
Toward on-site applications
Generally, assessment by the naked eye is considered the primary advantage of colour-changing test strips over fluorescence-changing test strips. However, today with cell phones being almost ubiquitously prevalent and utmost of these devices being equipped with a digital camera, we tested the present sensor matrix for a possible cell phone-based read-out. For this purpose, half of a strip was coated with 1-doped hydrogel and the other half with 2-doped hydrogel for internal referencing. After dipping into solutions containing various KOH concentrations, the strip was photographed (with the camera's LED flash always firing). A defined area (10 × 10 mm) of both halves of the strip in each image was analysed for their RGB values, which were then transformed to the CIE's XYZ scale. After averaging the latter values and plotting them versus pH, a calibration curve as that shown in Fig. 9 was obtained (see Fig. S6† for details). The good agreement of the data obtained by this simple on-site procedure, requiring only a conventional cell phone (an app allowing for direct data conversion and analysis in a smart phone is easily imaginable), with the fluorometer-based data shows the high potential such simple fluorescent sensors have in combination with rampant electronic communication devices.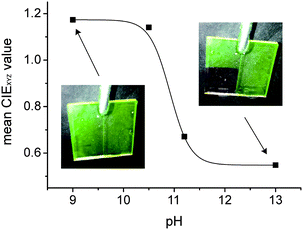 | ||
| Fig. 9 pH calibration curve obtained with half-1/half-2 coated test strips photographed with a cell phone camera after dipping into the respective KOH-containing solution. For details, see Fig. S6.† | ||
Conclusions
The development and characterization of a robust and reversible fluorescent pH sensing matrix coated onto a 3D epoxy polymer support for the alkaline pH range have been presented here. The embedded pH indicator dye, bearing a phenolic OH at the para-position of a meso-phenyl moiety attached to a BODIPY core, was synthesised through a simple one-pot synthesis procedure and shows high brightness independent of solvent polarity. The polymer material creates a microenvironment for the hydrophobic dye which is comparable to an aqueous ethanolic solution, as shown by fluorescence lifetime measurements, rendering the composite matrix immune against detrimental effects of water on the optical properties of 1 such as dye aggregation and low solubility. The fluorescence intensity of the film in water also remains unaffected upon addition of various anions and a high electrolyte background. With this approach the application of a rather hydrophobic dye could be expanded to aqueous media and a dipstick sensor for the alkaline region could be introduced. Due to the gigantic change in brightness by more than four orders of magnitude, the response of the sensor material upon a change in pH is also visible to the naked eye and can be assessed conveniently by use of a cell phone camera.Acknowledgements
This work was supported by the Innovationsfonds of BAM/BMWi. The authors thank Dr. P. Dietrich (BAM Div. 6.8 Surface Analysis and Interfacial Chemistry) for carrying out the XPS analysis and I. Feldmann (BAM Div. 4.2 Materials and Air Pollutants) for conducting the ESEM measurements. We are grateful to T. Fischer (BAM 1.9) for support with the fluorescence lifetime measurements and valuable discussions.Notes and references
- A. A. Belyustin, J. Solid State Electrochem., 2011, 15, 47–65 Search PubMed.
- J. N. Moorthy, T. Shevchenko, A. Magon and C. Bohne, J. Photochem. Photobiol., A, 1998, 113, 189–195 Search PubMed.
- H. Tomiyasu and Y. Asano, Prog. Nucl. Energy, 1998, 32, 421–427 Search PubMed.
- S. I. Stepanov, A. V. Boyarintsev, M. V. Vazhenkov, B. F. Myasoedov, E. O. Nazarov, A. M. Safiulina, I. G. Tananaev, H. V. So, A. M. Chekmarev and A. Y. Civadze, Russ. J. Gen. Chem., 2011, 81, 1949–1959 Search PubMed.
- T. Astrup, R. Jakobsen, T. H. Christensen, J. B. Hansen and O. Hjelmar, Waste Manage. Res., 2006, 24, 491–502 Search PubMed.
- W. M. Mayes, L. C. Batty, P. L. Younger, A. P. Jarvis, M. Koiv, C. Vohla and U. Mander, Sci. Total Environ., 2009, 407, 3944–3957 CrossRef CAS.
- P. B. Dhamole, R. R. Nair, S. F. D'Souza and S. S. Lele, Appl. Biochem. Biotechnol., 2008, 151, 433–440 Search PubMed.
- P. Sharma, H. Kaur, M. Sharma and V. Sahore, Environ. Monit. Assess., 2011, 183, 151–195 Search PubMed.
- W. M. Mayes, P. L. Younger and J. Aumonier, Environ. Sci. Technol., 2006, 40, 1237–1243 Search PubMed.
- V. O. Biederbeck, C. A. Campbell, H. Ukrainetz, D. Curtin and O. T. Bouman, Can. J. Soil Sci., 1996, 76, 7–14 Search PubMed.
- Q. Huang, Y. Peng, X. Li, H. F. Wang and Y. Z. Zhang, Curr. Microbiol., 2003, 46, 169–173 Search PubMed.
- A. Dayanandan, J. Kanagaraj, L. Sounderraj, R. Govindaraju and G. S. Rajkumar, J. Cleaner Prod., 2003, 11, 533–536 Search PubMed.
- C. Swaminathan, P. Pyke and R. F. Johnston, Miner. Eng., 1993, 6, 1–16 Search PubMed.
- M. Graefe, G. Power and C. Klauber, Hydrometallurgy, 2011, 108, 60–79 CrossRef.
- D. Joseph, Met. Finish., 2003, 215, 36–43 Search PubMed.
- K. Horikoshi, FEMS Microbiol. Rev., 1996, 18, 259–270 CAS.
- T. A. Krulwich, Mol. Microbiol., 1995, 15, 403–410 Search PubMed.
- A. S. Kocincova, S. Nagl, S. Arain, C. Krause, S. M. Borisov, M. Arnold and O. S. Wolfbeis, Biotechnol. Bioeng., 2008, 100, 430–438 CrossRef CAS.
- N. Boens, W. Qin, M. Baruah, W. M. De Borggraeve, A. Filarowski, N. Smisdom, M. Ameloot, L. Crovetto, E. M. Talavera and J. M. Alvarez-Pez, Chem.–Eur. J., 2011, 17, 10924–10934 Search PubMed.
- J. Han and K. Burgess, Chem. Rev., 2010, 110, 2709–2728 CrossRef CAS.
- Y. Chen, H. Wang, L. Wang, Y. Bian and J. Jiang, J. Org. Chem., 2011, 76, 3774–3781 CrossRef CAS.
- S. M. Borisov and O. S. Wolfbeis, Chem. Rev., 2008, 108, 423–461 CrossRef CAS.
- A. Safavi and H. Abdollahi, Anal Chim. Acta, 1998, 367, 167–173 CrossRef CAS.
- A. Safavi and M. Bagheri, Sens. Actuators, B, 2003, 90, 143–150 CrossRef.
- T. Werner and O. S. Wolfbeis, Fresenius' J. Anal. Chem., 1993, 346, 564–568 CrossRef CAS.
- H. W. Xu and O. A. Sadik, Analyst, 2000, 125, 1783–1786 RSC.
- T. A. Canada, D. B. Beach and Z. L. Xue, Anal. Chem., 2005, 77, 2842–2851 CrossRef CAS.
- A. Treibs and F. H. Kreuzer, Liebigs Ann. Chem., 1968, 718, 208–223 CrossRef CAS.
- T. Bura, P. Retailleau, G. Ulrich and R. Ziessel, J. Org. Chem., 2011, 76, 1109–1117 CrossRef CAS.
- N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130–1172 RSC.
- A. Coskun and E. U. Akkaya, J. Am. Chem. Soc., 2006, 128, 14474–14475 CrossRef CAS.
- H. Sunahara, Y. Urano, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2007, 129, 5597–5604 CrossRef CAS.
- H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620–2640 CrossRef CAS.
- H. Y. Lee, D. R. Bae, J. C. Park, H. Song, W. S. Han and J. H. Jung, Angew. Chem., Int. Ed., 2009, 48, 1239–1243 CrossRef CAS.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS.
- M. Kollmannsberger, K. Rurack, U. Resch-Genger and J. Daub, J. Phys. Chem. A, 1998, 102, 10211–10220 CrossRef CAS.
- U. Resch and K. Rurack, Proc. SPIE-Int. Soc. Opt. Eng., 1997, 3105, 96–103.
- Z. Shen, H. Rohr, K. Rurack, H. Uno, M. Spieles, B. Schulz, G. Reck and N. Ono, Chem.–Eur. J., 2004, 10, 4853–4871 CrossRef CAS.
- H. Röhr, C. Trieflinger, K. Rurack and J. Daub, Chem.–Eur. J., 2006, 12, 689–700 CrossRef.
- G. M. Sheldrick, SHELXS-97, Program for the Solution of Crystal Structures, Universität Göttingen, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL-97, Program for the Crystal Structure Refinement, Universität Göttingen, 1997 Search PubMed.
- B. M. Weidgans, C. Krause, I. Klimant and O. S. Wolfbeis, Analyst, 2004, 129, 645–650 RSC.
- ISO 15472, Int. Org. f. Standardization, Geneva, Switzerland, 2010.
- ISO 19318, Int. Org. f. Standardization, Geneva, Switzerland, 2004.
- G. Beamson and D. Briggs, High Resolution XPS of Organic Polymers, John Wiley & Sons, Chichester, UK, 1st edn, 1992 Search PubMed.
- C. Dumas-Verdes, F. Miomandre, E. Lepicier, O. Galangau, T. T. Vu, G. Clavier, R. Meallet-Renault and P. Audebert, Eur. J. Org. Chem., 2010, 2525–2535 Search PubMed.
- Y. Kubota, J. Uehara, K. Funabiki, M. Ebihara and M. Matsui, Tetrahedron Lett., 2010, 51, 6195–6198 CrossRef CAS.
- I. Pochorovski, B. Breiten, W. B. Schweizer and F. Diederich, Chem.–Eur. J., 2010, 16, 12590–12602 Search PubMed.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS.
- D. Tleugabulova, Z. Zhang and J. D. Brennan, J. Phys. Chem. B, 2002, 106, 13133–13138 CrossRef CAS.
- F. Bergstrom, I. Mikhalyov, P. Hagglof, R. Wortmann, T. Ny and L. B. A. Johansson, J. Am. Chem. Soc., 2002, 124, 196–204 CrossRef.
- J. Wang, Y. Hou, C. Li, B. Zhang and X. Wang, Sens. Actuators, B, 2011, 157, 586–593 Search PubMed.
- M. Maus and K. Rurack, New J. Chem., 2000, 24, 677–686 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC reference number 886601. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2an35860c |
| This journal is © The Royal Society of Chemistry 2013 |