Recent advances in fluorescent nucleic acid probes for living cell studies
Kemin
Wang
*,
Jin
Huang
,
Xiaohai
Yang
,
Xiaoxiao
He
and
Jianbo
Liu
State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Institute of Biology, Hunan University, Key Laboratory for Bio-Nanotechnology and Molecule Engineering of Hunan Province, Changsha 410082, China. E-mail: kmwang@hnu.edu.cn; Fax: +86 731 88821566; Tel: +86 731 88821566
First published on 19th October 2012
Abstract
Living cell studies can offer tremendous opportunities for biological and disease studies. Due to their high sensitivity and selectivity, minimum interference with living biological systems, ease of design and synthesis, fluorescent nucleic acid probes (FNAPs) have been widely used in living cell studies, such as for intracellular detection, cell detection, and cell-to-cell communication. Here, we review the general requirements and the recent developments in FNAPs for living cell studies. We broadly classify these designs as hybridization probes and aptamer probes. For hybridization probes, we describe recently developed designs, such as nanomaterial-based and amplification-based hybridization probes. For aptamer probes, we discuss four general paradigms that have appeared most frequently in the literature: nanomaterial-based, nanomachine-based, cell surface-anchored and activatable aptamer probe designs in vivo. FNAPs promise to open up new and exciting opportunities in biological marks detection for a wide range of biological and medical applications.
 Kemin Wang | Kemin Wang is a professor of Chemistry and Biomedical Engineering at Hunan University, since 1992. He obtained a PhD degree in analytical chemistry at Hunan University in 1987 and carried out post-doctoral studies at the Union High Institute of Technology of Zurich in Switzerland from 1989 to 1991. His research interests mainly focus on biological analytical chemistry at nanometer and single molecule level. |
 Jin Huang | Jin Huang received his PhD in analytical chemistry at Hunan University in 2011. From 2009–2011, he studied as a visiting student at the University of Florida, USA. Currently, he is a postdoctoral researcher in chemical engineering at Hunan University. His research interests focus on DNA probe engineering for bio-chemical analysis. |
 Xiaohai Yang | Xiaohai Yang received his PhD in analytical chemistry at Hunan University in 2000 and carried out post-doctoral studies at the University of Tokyo from 2002 to 2004. He is a professor of chemistry in Hunan University. His research interests are in the field of optical biosensing at nanometer and single molecule level. |
 Xiaoxiao He | Xiaoxiao He is a professor in the Biological School of Hunan University, since 2006. She received her PhD in analytical chemistry at Hunan University in 2003. From 2009 to 2010, she studied in Stanford University, USA, as a visiting scholar. Her research interests are in the field of bio-chemical analysis and bio-nanotechnology. |
 Jianbo Liu | Jianbo Liu received his PhD in analytical chemistry at Hunan University in 2011. Since then he has worked as an assistant professor of Chemistry at Hunan University. His research interests are fluorescent nanomaterials and single-molecule imaging. |
1. Introduction
The cell, as a functional basic unit of life, concentrates the arcane of life in a microcosmic world. Professor X. Sunney Xie of Harvard University has compared living cells to chemists' test tubes.1 It has been said that living cell studies may help humans understand themselves, explore the mysteries of life, elucidate disease mechanisms, and help provide methods to cure diseases. Particularly, intracellular biomolecular interaction studies have been widely considered essential for advancement in molecular biology, disease pathophysiology, drug discovery, diagnostic and therapeutic applications. However, the milieu of the living cell is different from that in a test tube environment. For example: (1) many biomolecules exist in low copy numbers and participate in stochastic reaction events in the cell that are hidden in test tubes with large numbers of molecules; (2) reactions often take place at a nonequilibrium steady state in the cell; (3) many reactions are coupled in the cell, resulting in networks of complex interactions. Therefore, all these special circumstances require higher sensitivity and resolution in time and space.Recently, fluorescent molecular probes have shown good potential in a wide range of applications that require sensitive detection of biological analytes in living biological systems.2–5 Fluorescent molecular probes are aimed to rationally design and synthesize molecules with specific properties, which might include target recognition and signal transduction.6–8 Ideally, the target recognition elements should have high affinity, high specificity, fast response and good generality for detecting a broad range of analytes. Signal transduction elements are responsible for converting molecular recognition events into physically detectable fluorescent signals such as intensity, lifetime or anisotropy. Of the various potential candidate molecules for rational design, which include small organic molecules, peptides and proteins, nucleic acids are the easiest to design and engineer.9,10
Nucleic acids, especially DNA bases, have been chosen as ideal building blocks for molecular engineering of probes for a variety of unique applications in living biological systems. In summary, the following may make nucleic acids engineering relatively easy and universal: (1) an understanding of Watson–Crick base pairing rule and the availability of structure prediction software make the design rational and smart; (2) the robotization of chemical synthesis with extreme accuracy and reproducibility assures no variation in production; (3) various nucleic acids related tool enzymes make it possible to optionally cut, ligate or prolong; (4) the skilled use of modified methods provide nucleic acids equipped with a variety of reporter groups and enable coupling to many different carriers, surfaces, nanoparticles, or other biomolecules; (5) with a combinatorial technology called SELEX (systematic evolution of ligands by exponential enrichment), it is possible to evolve nucleic acids to bind to a wide range of targets, such as metal ions, small organic molecules, drugs, proteins, and even whole cells with high affinity and specificity, and these binding nucleic acids are known as aptamers.11–16
The application of aptamers increasingly enables the fields of biosensing, diagnostics and therapeutics to be explored. There have been many excellent reviews in recent years with different emphases.17–20 The introduction of aptamers serves as an excellent recognition element and makes nucleic acids molecular engineering more flexible and multifunctional. In a general approach, these nucleic acid probes are designed to have a fluorescence reporter group covalently attached to an oligonucleotide sequence, which serves as a recognition element for target binding. Without the target, the reported fluorescence signal retains one state. Once a target is added the nucleic acid probes binds to it and produces a distinctive fluorescence signal.
In this review, we discuss the general requirements of fluorescent nucleic acid probes (FNAPs) for living cell studies, and we then introduce recently designed FNAPs within the last five years. We broadly classify these designs as hybridization probes and aptamer probes, and then generally describe the rational design schemes of each, as well as discussing in detail specific examples of each.
2. General requirements of FNAPs for living cell studies
There are several general requirements for successful FNAPs in living cell studies, including delivery, stability, targeting and detectability.2.1. Delivery
To measure intracellular level of molecules, the probes must be introduced into cells through the plasma membrane, which is quite lipophilic and restricts the transport of various molecules. Nucleic acids probes, as hydrophilic molecules, cannot freely traverse plasma membranes. Up to now, several techniques have been used to deliver probes across the plasma membrane barrier, including microinjection,21–23 transfection with cationic lipids,24 the use of cell-penetrating peptides (CPP),25–27 streptolysin O (SLO),28,29 and nanomaterials.30–32 Among these, microinjection is effective and yields high-quality data, but it is tedious and can be used to deliver probes to only a few cells. The other techniques, in contrast, can be used to deliver probes into a large number of cells.2.2. Stability
The stability of probes plays an important role in avoiding false positive signals and lowering background signals. Nucleic acids probes with nature backbones are degraded rapidly inside living cells because various enzymes in the cell have the ability to degrade the nature backbones of oligonucleotides. It is reported that the half life of molecular beacons is around 30 minutes due to endogenous nuclease degradation.33 Thus, it is desirable to enhance the stability of probes when considering living cells. Synthesizing probes from modified nucleotides such as 2′-O-methylribonucleotides22 and locked nucleic acids34 showed improved stability inside the cells. However, it is essential to be aware that backbone modifications to nucleic acid probes can alter the thermodynamics of probe hybridization. For example, the presence of 2′-O-methylribonuclotides and locked nucleic acids linkages increases a probe's affinity for its target. Another strategy is to use nanomaterials as probe carriers which can enhance nuclease resistance, since it has been previously reported that immobilization or adsorption of oligonucleotides on nanomaterial surface provides stability in the presence of enzymes.35–38 Materials frequently used contain gold nanoparticles, silica nanoparticles, carbon nanotubes, graphene oxide and so forth. For example, Mirkin et al. revealed that the conventional probe was degraded approximately 4.5 times more rapidly than when immobilized on gold nanoparticles.372.3. Targeting
The probe must interact selectively with a target molecule or other structure. As an example, when probes for mRNA imaging are introduced rapidly into the nucleus, they are held there by nucleic acid binding proteins that are abundant in the nucleus, therefore there is no time for them to bind to cytoplasmic mRNAs. On the other hand, the target sequence in mRNA may not be available for the binding of a probe owing to the presence of a secondary structure in the RNA. Thus, it is usual to select the loop of the mRNA as the target region because it is more accessible than the stem. It is reported that only 5% of all possible oligonucleotide probes efficiently bind their complements in a full length mRNA.39 The RNA fold WebServer (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi), freely available on the internet may improve the odds of designing successful probes.402.4. Detectability
The fluorescence output of the probes must be compatible with the instrumentation that is to be used for detection. We think at least three factors (wavelength, brightness and photobleaching) might influence the detectability of the probes.Wavelength of the fluorophores is the first issue to affect the probe detectability. It is known that fluorophores require light to emit light. However, excitation in the ultraviolet can cause tissue damage;41 blue and green excitation light have poor tissue penetration; yellow and red light lead to excessive autofluorescence because the bulk of naturally occurring endogenous fluorophores, mostly hemoglobin and related molecules, are also excited in this range.42 To maximize fluorescence signals, the optimal excitation wavelength of a fluorophore is in the deep red or near-infrared range (650–900 nm) because of the combined virtues of good tissue penetration and low autofluorescence.43,44 When considering the Stocks shift, the selection of both excitation and emission wavelengths are usually in the deep red or near-infrared range. Brightness of the fluorophore is the second consideration, because of that the brighter the agent, the more sensitive the probes. Usually, the brightness is proportional to the fluorescence quantum yield, which is the ratio of photons emitted through fluorescence to photos absorbed. For example, the fluorescence quantum yield of Cy5 is 0.28 and Cy3 is 0.04 in PBS buffer. However, if the solvent is changed to ethanol, the Cy5 is about 0.40, and the Cy3 is about 0.09 in ethanol.45 Thus, it should be noted that fluorescence quantum yields are often very sensitive to environmental factors (such as pH and solvent).46 Additionally, under the high-intensity illumination conditions used for fluorescence microscopy, the irreversible destruction of the excited fluorophore (photobleaching) often becomes the factor limiting fluorescence detectability.46–48 Fortunately, it has been found that, for example, quantum dots and dye doped core–shell nanoparticles which resist photobleaching effectively can in part solve the problem.49–51
3. Hybridization probes for living cell studies
Hybridization probes are usually used to image endogenous mRNAs. Only when the probes hybridize to their target RNA can the fluorescent signal be detected, normally this is done by using label moieties that interact either by fluorescent resonance energy transfer (FRET) or collisional quenching. These hybridization probes are divided into five types by different designs: competitive hybridization probes,52,53 side-by-side probes,54,55 quenched autoligation probes,56,57 molecular beacon probes58,59 and dual molecular beacon FRET probes60 (Fig. 1). Bao and Tyagi's reviews2–4 have basically described well the development of hybridization probes in living cells. Therefore, herein, we will omit discussion of the above classical probes and instead discuss recent new advances.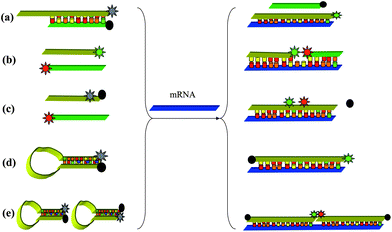 | ||
| Fig. 1 Hybridization probes for intracellular mRNA detection. (a) Competitive hybridization probes, (b) side-by-side probes, (c) quenched autoligation probes, (d) molecular beacon probes, (e) dual molecular beacon FRET probes. | ||
3.1. Nanomaterial-based hybridization probe designs
As dicussed in sections 2.1 and 2.2, the probes often require additional agents for cellular internalization, and can be unstable in cellular environments. These factors can lead to a high background signal and the inability to detect targets. However, the unique properties of some nanomaterials might have the potential to solve these problems. For example, gold nanoparticles (AuNPs), carbon nanotubes (CNTs) and graphene oxide (GO).AuNPs are important nanomaterials, which typically have dimensions ranging from 1–100 nm. In recent years, AuNPs have led to new and exciting developments with enormous potential in biology and medicine.61–63 For example, Mirkin et al. proposed a novel nano-flares probe, which took advantage of the high efficient fluorescent quenching properties of gold, cellular uptake of oligonucleotide nanoparticle conjugates without the use of transfection agents, and the enzymatic stability of such conjugates, thus overcoming many of the challenges in creating effective intracellular probes (Fig. 2(a)).37,64 Specifically, AuNPs were functionalized with thiolated oligonucleotides containing an 18-base recognition element to a specific RNA transcript via gold thiol bond formation. Oligonucleotide-functionalized AuNPs were then allowed to hybridize with short Cy5 dye-terminated reporter sequences capable of acting as “flares” when displaced by a longer target or target region. In the bound state, the Cy5 fluorescence of the reporter strand is quenched due to proximity to the AuNP surface. In the presence of a target, the flare strand is displaced and liberated from the AuNP by forming the longer and more stable duplex between the target and the oligonucleotide-modified AuNP.
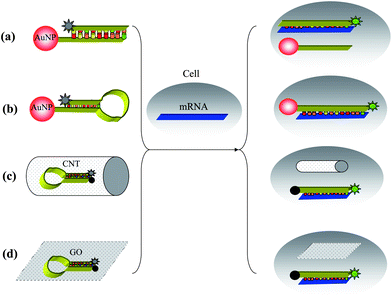 | ||
| Fig. 2 Strategies for living cell imaging of mRNA using (a) nano-flares, (b) hairpin DNA-coated AuNP, (c) probes–CNTs complex and (d) MBs–GO complex. | ||
Wright et al. presented a strategy for living cell imaging of mRNA using hairpin DNA-coated gold nanoparticle (hAuNP). In this approach, a modification of a molecular beacon-based strategy, a hairpin DNA moiety (h) is coupled to a AuNP surface. The hairpin DNA structure consists of a 5′-thiol followed by a 10-base polythymine linker sequence and a stem-loop-stem sequence, and is terminated at the 3-end with a fluorophore. The loop region is designed to hybridize with the target RNA of interest. In the closed state, the hairpin conformation is maintained by the complementary 5′ and 3′ 5-base stem sequences. Additionally, in this closed state, the 5′-thiol group enables linkage to the AuNP surface with sufficient proximity to permit AuNP quenching of fluorescence from the 3′ dye. Specific hybridization of hAuNP with target RNA opens the hairpin DNA sequence, which then separates the 3′ fluorophore from the 5′ AuNP at a sufficient distance to overcome fluorescence quenching. Besides the advantages of nano-flares, hAuNP once bound to target mRNA sequence, remains hybridized to the target, enabling spatial and temporal studies of trafficking and downstream analysis (Fig. 2(b)).65,66
CNTs are also important nanomaterials, which have already been used in many fields because of its excellent biocompatibility, cellular permeability, effective endocytosis, fluorescence quenching ability, as well as its strong adsorption.67–70 Tan et al. have developed CNTs as carriers for ssDNA probes delivery for mRNA detection in living cells.71 The specific DNA probes were first labeled with Cy3 and complexed with CNTs. The CNTs–probes complex showed increased fluorescence upon binding manganese superoxide dismutase (MnSO) mRNA. The MnSO probes/CNTs complex can be delivered by incubation with MDA-MB-231 breast carcinoma cells. Under normal culture conditions, the cell line has a low MnSO expression level; however, when exposed to lipopolysaccharide (LPS), an inflammatory mediator involved in Escherichia coli. Bacterial sepsis, MnSOD mRNA expression levels increase substantially. As a result of its resistance to enzymatic cleavage, the MnSO probes/CNTs complex showed a low fluorescence background signal before the cells were stimulated with LPS. However, following LPS stimulation, the complex was still functional after long intracellular incubation times, and hybridization with target mRNA sequences produced a high fluorescent signal which could be detected by confocal microscopy (Fig. 2(c)).
GO, which is a water-soluble derivative of graphene, has attracted increasing interest in biological applications because of its unique characteristics, including good water dispersion, facile surface modification and high mechanical strength.72–76 Moreover, GO is a good nucleic acid adsorption material and a good energy acceptor in energy transfer due to its superior electronic properties.77–79 Yang and co-workers reported that functional GO sheets are able to protect molecular beacon (MB) from cleavage and can deliver MB into cells. MB is a hairpin-shaped DNA with a self-complementary stem that brings a terminal-labeled fluorophore and a quencher into close proximity, causing fluorescence of the fluorophore to be quenched by energy transfer. When a MB hybridizes with its complementary target, the beacon undergoes a spontaneous conformational reorganization with the opening of the stem, leading to fluorescence restoration.7 When the cells were treated with the MB–GO complex, a high fluorescence signal was observed for most of the cells, while a lower fluorescence signal was detected when the cells were treated with free MB probe and control MB–GO complex under the same conditions (Fig. 2(d)).38
Nanomaterial-based probes represent newly effective intracellular mRNA imaging tools. These versatile intracellular probes maintain advantages over traditional molecular mRNA imaging techniques in that they circumvent the use of transfection or cell penetrating agents, and they are resistant to nuclease degradation.
3.2. Amplification-based hybridization probe designs
Amplification strategies have often been used to combine hybridization probes for sensitive nucleic acids detection. These methods attract substantial efforts due to possible applications in medical diagnosis, detection of environmental pollutants, controlling food quality, and forensic analyses. Because the nucleic acids of interest may be present in very small amounts, it is necessary to develop amplification techniques that enable the detection of trace levels of a specific sequence. Here, we focus on the application of amplification strategies for intracellular mRNA detection. For example, Pierce et al. introduced the concept of hybridization chain reaction (HCR) in 2004, in which stable DNA monomers assemble only upon exposure to a target DNA fragment. In the simplest version of this process, two stable species of DNA hairpins coexist in solution until the introduction of initiator strands triggers a cascade of hybridization events that yields nicked double helices analogous to alternating copolymers.80 This functionality allows it to act as an amplifying transducer for biosensing applications. We successfully combined HCR and pyrene molecules for highly sensitive DNA detection.81 Willner et al. combined HCR with DNAzyme to detect very low concentrations of target.82,83 As a real example of mRNA detection in living cells, Pierce et al. report a multiplexed fluorescent in situ hybridization method based on orthogonal amplification with HCR.84 With this approach, RNA probes complementary to mRNA targets trigger chain reactions in which fluorophore-labled RNA hairpins self-assemble into tethered fluorescent amplification polymers. The programmability and sequence specificity of these amplification cascades enable multiple HCR amplifiers to operate orthogonally simultaneously in the same sample (Fig. 3).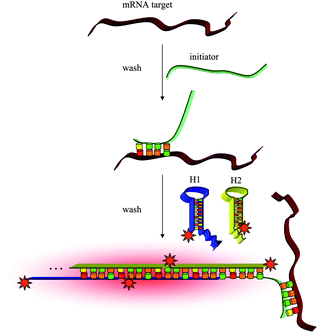 | ||
| Fig. 3 In situ hybridization using fluorescent HCR amplification. | ||
Robust performance is achieved when imaging five target mRNAs simultaneously in fixed whole-mount and sectioned zebrafish embryos (Fig. 4). It seems that this approach is potentially suited for use in a variety of biological contexts including fixed cells, embryos, tissue sections and micropopulations. By coupling HCR initiators to aptamer or antibody probes, HCR amplification is also potentially suitable to multiplex imaging of small molecules and proteins. Future work is required to explore these possibilities.
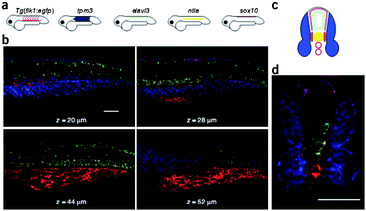 | ||
| Fig. 4 Multiplexed imaging in fixed whole-mount and cross-sectioned zebrafish embryos. (a) Expression atlas for five target mRNAs. (b) mRNA expression imaged using confocal microscopy at four planes within an embryo. (c) Expression atlas for five target mRNAs (anterior view). (d) mRNA expression imaged within a 200 μm zebrafish section using confocal microscopy. Adapted from ref. 84. Reprinted by permission from Nature Publishing Group, copyright 2009. | ||
4. Aptamer probes for living cell studies
Aptamers are single-stranded DNA or RNA oligonucleotides selected from an in vitro method known as SELEX, as originally developed by Gold, Szostak et al. in 1990.11–13 Aptamers have been selected for a wide range of target molecules, from ions and small molecules to proteins and even whole living cells. In addition to high affinity and selectivity, aptamers possess several advantages over antibodies. These include the ease of synthesis, stability under room conditions, lack of immunogenicity, rapid tissue penetration, and ease of modification.85,86 Therefore, aptamers have great potential as molecular probes for biomedical applications. However, exploration of aptamers for intracellular analysis and in situ monitoring is still in its early stage.4.1. Nanomaterial-based aptamer probe designs
Adenosine triphosphate (ATP) is considered as an energy carrier in cells, and the basis of biological activity. Mirkin et al. described the development of novel aptamer-nanoparticle probes, termed aptamer nano-flares, which are designed to detect and quantify the intracellular analyte ATP. These nanostructures consist of AuNP that is functionalized with ATP aptamers hybridized to Cy5 labeled flares (Fig. 5(a)). In the bound state, the fluorescence of a flare strand is quenched by AuNP. In the presence of ATP target molecules, ATP binds to the aptamer causing a conformational change and results in a newly folded second structure. This folded structure disrupts the Watson–Crick base-pairing between the aptamer and the flare, which causes flares to be liberated with an increase in fluorescence due to the greater distance of flare from the gold surface. When HeLa cells were incubated with the aptamer nano-flares and imaged using scanning confocal microscopy, HeLa cells treated with aptamer nano-flares were highly fluorescent when compared to those treated with the mismatched sequence.87 Lin and coworkers demonstrated cellular delivery and in situ molecular probes in living cells by employing graphene oxide nanosheets (GO-nS) as DNA cargo and sensing platforms (Fig. 5(b)). Significant FAM fluorescence could be observed for JB6 cells (mouse epithelial cells) incubated with aptamer-FAM/GO-nS, but not in random DNA-FAM/GO-nS, indicating successful intracellular aptamer delivery and ATP probing in living cells.88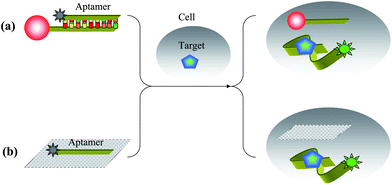 | ||
| Fig. 5 Schematic illustrations of intracellular ATP detection by (a) aptamer nano-flares and (b) aptamer–GO complex. | ||
With the rapid development of Cell-SELEX, a variety of aptamers have been selected for several types of cancer cells, including lymphocytic leukemia, myeloid leukemia, liver cancer, small-cell lung cancer and non-small-cell lung cancer.15,16,89–91 These aptamers all showed high affinity and excellent specific selectivity against their target cells. A feasible strategy for detecting cancer in its early stages must develop an effective method for detecting cancer cells in low abundance. In recent years, several methods have been developed to achieve this goal. Because aptamers are excellent recognition elements, they can also be conjugated to fluorescent molecules for targeted imaging of the cells, or to nanoparticles for signal enhancement. For example, in 2006, Tan et al. described aptamer-conjugated nanoparticles (ACNPs) for the rapid detection of acute leukemia cells, which use high-affinity DNA aptamers for recognition. An 88-nt oligonucleotide sequence with specific binding properties for CCRF-CEM acute leukemia cells was attached to both magnetic nanoparticles (MNPs) and fluorescent nanoparticles (FNPs) to develop a specific platform for collecting and imaging intact leukemia cells from mixed cell and whole blood samples.92 In 2007, they further developed ACNP protocols to perform extraction of multiple cancer targets using additional high-affinity DNA aptamers for cognition. Three different cell samples, CEM, Ramos, and Toledo cells, were stepwise extracted and detected by fluorescent imaging using MNPs and FNPs modified by three aptamers (Fig. 6).93
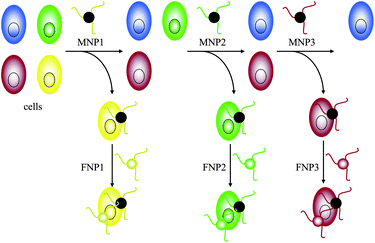 | ||
| Fig. 6 Schematic representation of three different cell samples stepwise extracted and detected by MNPs and corresponding FNPs. | ||
In 2009, Kang et al. demonstrated multiplex detection of cancer cells using quantum dots (QDs) conjugated aptamers. Three aptamers (TTA1, AS1411, and MUC-1) were conjugated to QDs with emission wavelengths of 605, 655, and 705 nm, respectively. Each of them showed specific fluorescent signal by confocal analysis.94 In 2011, our group reported a one-step engineering of intrinsically fluorescent silver nanoclusters (AgNCs)–aptamer assemblies that allows the development of facile and specific luminescent labels for target tumor cells recognition and analysis (Fig. 7).95 Here, AgNCs have been utilized as signal moieties because of their outstanding spectral features and biocompatibility. A sgc8c-AgNCs assembly showed specific binding to target CEM cells, not Ramos cells. We predict that this strategy holds great potential for use in cancer cell analysis or clinical diagnosis when more aptamers have been selected against tumor cells.
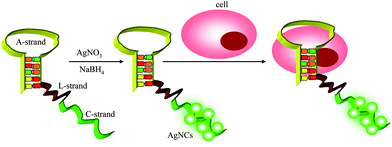 | ||
| Fig. 7 Schematic illustration of an one-step AgNCs–aptamer assembly as a luminescent label for tumor cells. | ||
4.2. Nanomachine-based aptamer probe designs
pH plays an important role in cellular processes, such as endocytosis, which show characteristic acidification profiles that are integral to endosome maturation, vesicle and cargo functions such as sorting of secretory molecules, growth factors, nutrients and toxins.96 As early endosomes they undergo a characteristic change of pH ranging from 6 to 6.2 in early endosomes, to pH 5.5 in late endosomes and pH 5 in lysosomes.97 Krishnan et al. presented the first example of nanoscale DNA nanomachine response to protons within a living cell.98 In this study, the I-switch consists of three oligonucleotides O1, O2 and O3, where O1 and O2 are hybridized onto sites adjacent to O3, leaving a one-base gap. O1 and O2 have single-stranded cytosine-rich overhangs designed such that each overhang forms one-half of a bimolecular i-motif. At acidic pH, these overhangs are protonated and the assembly can fold to form an intramolecular i-motif (Fig. 8). This DNA nanodevice has cytosine-rich regions that function as sensors for pH. At acidic pH, the DNA structure is protonated and the assembly can fold to form an intramolecular i-motif, thus bringing the Alex-488 and Alex-647 labels at the 3′ and 5′ termini into proximity, and which show FRET signal. The I-switch can be used to visualize pH changes in real time; where a sharp decrease in donor/acceptor ratio (D/A) is observed over a period of 30 min followed by a slower decrease over 2 h, this suggests that early endosomes in this pathway rapidly acidify to form the late endosome, which then slowly matures to the lysosome. Thus the I-switch reports spatio-temporal pH changes efficiently where variation in pH correlates well to molecular processes associated with endosome maturation.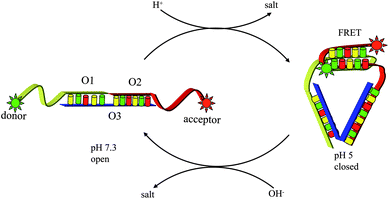 | ||
| Fig. 8 Spatial and temporal mapping of pH changes during endocytosis using the I-switch in living cells. | ||
Furthermore, Krishnan et al. chose it as in vivo system to explore the functionality of the DNA nanomachine I-switch. It was demonstrated that post endocytosis the I-switch is functional recapitulating its pH-sensing properties in vivo, with high fidelity, and the highest dynamic range is observed at pH 5.3–6.6 (Fig. 9). In this study, for the first time, autonomous functionality of a rationally designed, synthetic DNA nanomachine in a living organism was demonstrated.99
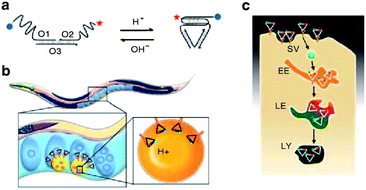 | ||
| Fig. 9 (a) Structure and working of I-switch. (b) I-switch uptake in coelomocytes postinjection in C. elegans. (c) Stages in endosomal maturation of receptor-mediated endocytosis of a ligand (SV, spherical vesicle; EE, early endosome; LE, late endosome; LY, lysosome). Adapted from ref. 99. Reprinted by permission from Nature Publishing Group, copyright 2011. | ||
4.3. Cell surface-anchored aptamer probe designs
Monitoring cell functions and cell-to-cell communication in the cellular environment has enormous implications for cell biology and regenerative medicine. Recently, Karp et al. presented a new application of a fluorescent aptamer sensor attached to a cell membrane, which was able to detect signaling molecules in the cellular environment.100 The sensor is an aptamer that binds to platelet-derived growth factor (PDGF) and contains a dye FAM and a quencher Dabcyl. When bound to PDGF, the aptamer changes conformation and the dye moves closer to the quencher, which decreases the fluorescent signal. The sensor, which is covalently attached to the membranes of mesenchymal stem cells (MSCs), can quantitatively detect, with high spatial and temporal resolution, PDGF that is added in the cell culture medium or secreted by neighbouring cells (Fig. 10). Imaging was recorded immediately after the PDGF was added to sensor-modified MSCs from the top/left (arrow shows direction) using a pipette tip. Furthermore, cell surface sensors were subsequently applied in mouse bone marrow by 24 h post-transplantation, the success of which suggested that nucleic-acid-sensor conjugated fluorophores are functional on the cell surface for at least a day under physiological conditions.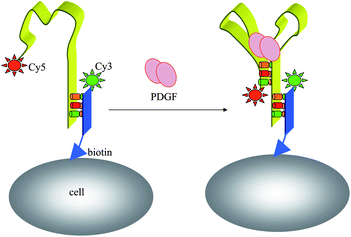 | ||
| Fig. 10 Fluorescent imaging of a cell-surface aptamer sensor for PDGF in cellular environments. | ||
Subsequently, Tokunaga et al. successfully utilized tocopherol-labeled aptamer direct anchoring to the cell surface for real-time monitoring of release of adenine compounds as a glio-transmitter, which was synchronized with calcium wave propagation in neighboring cells.101 We conclude that this current work expands the research field of aptamer sensors and has provided a novel design idea.
4.4. Activatable aptamer probe designs in vivo
Molecular imaging, which has been defined as in vivo characterization and measurement of biological process at the cellular and molecular levels, is widely considered the future for medical imaging.5 FNAPs have attracted increasing attention in in vivo applications due to their various virtues. However, exploration of FNAPs in vivo still remains in its infancy. To address the limitation of current molecular probes, like poor sensitivity and low specificity, we first report fluorescent aptamer probes for tumor imaging in a mouse.102 Using Cy5-TD05 as the probe, in vivo efficacy of aptamer-based molecular imaging was tested in Ramos xenograft nude mice. After intravenous injection of Cy5-TD05 into mice bearing grafted tumors, noninvasive, whole-body fluorescence imaging allowed spatial and temporal distribution to be directly monitored. The results showed that the aptamers can effectively recognize tumors with high sensitivity and specificity, thus establishing the efficacy of fluorescent aptamers for diagnostic applications and in vivo studies requiring real-time molecular imaging. However, because always-on aptamer probes have constant signals, image contrasting is critically limited by a high background. A cancer site can be clearly observed only after physiological clearing of unbound aptamers, which leads to a long diagnosis time, which is further compromised by substantial consumption of bound aptamers. Considering the above issues, we have designed a novel activatable aptamer probe (AAP) with a fluorophore and a quencher attached at either terminus. In the absence of a target, the AAP is hairpin structured, resulting in a quenched fluorescence. When the probe is bound to membrane receptors of the target cancer cell, its conformation is altered, causing an activated fluorescence signal (Fig. 11).103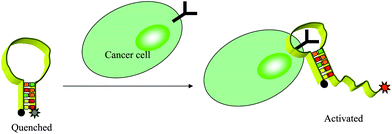 | ||
| Fig. 11 Schematic representation of the novel strategy for in vivo cancer imaging using AAP based on cell membrane protein triggered conformation alteration. | ||
As expected, AAP can be activated by target cancer cells with faster restoration of fluorescence achieved in the tumor site compared to other areas. Unlike AAP, the always-on probes suffered from a high fluorescence background in the whole body from an early time point (Fig. 12). Considering the expansion of aptamer discovery for varying cancer targets, the developed AAP strategy described may hold great potential as a versatile molecular probe for in vivo cancer imaging with high sensitivity and specificity.
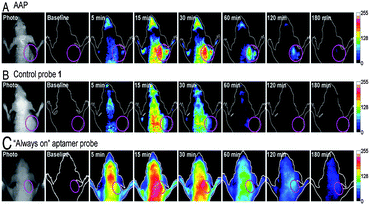 | ||
| Fig. 12 In vivo time-dependent fluorescence imaging of CEM tumors. CEM tumor-bearing nude mice were intravenously injected with (A) AAP, (B) control probe 1, and (C) the “always-on” aptamer probe, respectively. Adapted from ref. 103. Reprinted by permission from National Academy of Sciences, USA, copyright 2011. | ||
5. Conclusions and outlook
In this review, we have described recent advances of FNAPs in living cell studies, particularly those occurring during the previous five years. From the general requirements of probes, to paradigms that have appeared most frequently in the literature, current state-of-the-art already enables imaging of FNAPs in many biological situations. It has been shown that increased demand for developments within molecular biology and medical applications has resulted in active research within the field. Although most of the challenges described for these probes have been efficiently addressed by the research introduced in this review, further improvements are needed to achieve greater levels of sensitivity, selectivity and practicability. Overall, research in this area still remains in its very early stages.Further areas of study are as following: (1) chemical modification of nucleic acids probes requires further development. (2) Integrated with current nanotechnologies and other sophisticated devices, FNAPs provide improved sensitivity and practicability in the detection of target molecules. (3) We predict that future research will involve unusual and ingenious schemes or principles to smartly detect targets by simpler methods. (4) Aptamer probes serve as the majority of nucleic acid probes, which act to expand the targets’ range of detection. Successful applications of nucleic acid probes in biological imaging depends greatly on SELEX techniques. More specific aptamers for different targets is desirable. (5) Understanding aptamer–target binding interactions is expected to lead to improved utilization of aptamers in living cell studies.
In addition, the particular in vivo system, such as mouse, zebrafish embryos and Caenorhabditis elegans, are promising test beds for FNAPs. These FNAPs can be designed to interrogate a variety of processes and to track molecularly complex phenomena thus opening up immense possibilities towards successful application of these probes in whole organism studies.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (90606003 and 21190040), National Basic Research Program (2011CB911002), International Science and Technology Cooperation Program of China (2010DFB30300), Program for Changjiang Scholar and Innovative Research Team in University, Program for New Century Excellent Talents in University (NCET-09-0338), and Hunan Natural Science Foundation (10JJ7002).Notes and references
- X. S. Xie, J. Xu and W. Y. Yang, Science, 2006, 312, 228–230 CrossRef CAS.
- P. Santangelo, N. Nitin and G. Bao, Ann. Biomed. Eng., 2006, 34, 39–50 CrossRef.
- W. J. Rhee and G. Bao, BMC Biotechnol., 2009, 9, 30–39 CrossRef.
- S. Tyagi, Nat. Methods, 2009, 6, 331–338 CrossRef CAS.
- H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620–2640 CrossRef CAS.
- A. A. Marti, S. Jockusch, N. Stevens, J. Ju and N. J. Turro, Acc. Chem. Res., 2007, 40, 402–409 CrossRef CAS.
- K. Wang, Z. Tang, C. J. Yang, Y. Kim, X. Fang, W. Li, Y. Wu, C. D. Medley, Z. Cao, J. Li, P. colon, H. Lin and W. Tan, Angew. Chem., Int. Ed., 2009, 48, 856–870 CrossRef CAS.
- J. Liu, Z. Cao and Y. Lu, Chem. Rev., 2009, 109, 1948–1998 CrossRef CAS.
- K. Sefah, J. A. Phillips, X. Xiong, L. Meng, D. V. Simaeys, H. Chen, J. Martin and W. Tan, Analyst, 2009, 134, 1765–1775 RSC.
- D. Li, S. Song and C. Fan, Acc. Chem. Res., 2010, 43, 631–641 CrossRef CAS.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CAS.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS.
- D. L. Robertson and G. F. Joyce, Nature, 1990, 344, 467–468 CrossRef CAS.
- P. L. Sazani, R. Larralde and J. W. Szostak, J. Am. Chem. Soc., 2004, 126, 8370–8371 CrossRef CAS.
- D. Shangguan, Y. Li, Z. Tang, Z. Cao, H. Chen, P. Mallikaratchy, K. Sefah, C. Yang and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11838–11843 CrossRef CAS.
- Z. Tang, D. Shangguan, K. Wang, H. Shi, K. Sefah, P. Mallikaratchy, H. Chen, Y. Li and W. Tan, Anal. Chem., 2007, 79, 4900–4907 CrossRef CAS.
- E. J. Cho, J. W. Lee and A. D. Ellington, Annu. Rev. Anal. Chem., 2009, 2, 241–264 CrossRef CAS.
- A. D. Keefe, S. Pai and A. D. Ellington, Nat. Rev. Drug Discovery, 2010, 9, 537–550 CrossRef CAS.
- X. Fang and W. Tan, Acc. Chem. Res., 2010, 43, 48–57 CrossRef CAS.
- A. B. Iliuk, L. Hu and W. A. Tao, Anal. Chem., 2011, 83, 4440–4452 CrossRef CAS.
- J. P. Leonetti, N. Mechti, G. Degols, C. Gagnor and B. Lebleu, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 2702–2706 CrossRef CAS.
- D. P. Bratu, B. J. Cha, M. M. Mhlanga, F. R. Framer and S. Tyagi, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13308–13313 CrossRef CAS.
- D. Y. Vargas, A. Raj, S. A. Marras, F. R. Framer and S. Tyagi, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17008–17013 CrossRef CAS.
- M. M. Mhlanga, D. V. Fung, F. R. Kramer and S. Tyagi, Nucleic Acids Res., 2005, 33, 1902–1912 CrossRef CAS.
- N. Nitin, P. J. Santangelo, G. Kim, S. Nie and G. Bao, Nucleic Acids Res., 2004, 32, e58 CrossRef.
- K. Melikov and L. V. Chernomordik, Cell. Mol. Life Sci., 2005, 62, 2739–2749 CrossRef CAS.
- H. Y. Yeh, M. V. Yates, A. Mulchandani and W. Chen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17522–17525 CrossRef CAS.
- P. J. Santangelo, B. Xie, A. Tsourkas and G. Bao, Nucleic Acids Res., 2004, 32, e57 CrossRef.
- A. K. Chen, M. A. Behlke and A. Tsourkas, Nucleic Acids Res., 2008, 36, e69 CrossRef.
- D. A. Giljohann, D. S. Seferos, W. L. Danile, M. D. Massich, P. C. Patel and C. A. Mirkin, Angew. Chem., Int. Ed., 2010, 49, 3280–3294 CrossRef CAS.
- P. Cheng, W. Hu, Y. Zhou, C. Fan and Q. Huang, Small, 2010, 6, 1686–1692 CrossRef.
- R. Hu, X. Zhang, R. Kong, X. Zhao, J. Jiang and W. Tan, J. Mater. Chem., 2011, 21, 16323–16334 RSC.
- T. L. Fisher, T. Terhost, X. Cao and R. W. Wang, Nucleic Acids Res., 1993, 21, 3857–3865 CrossRef CAS.
- Y. Wu, C. J. Yang, L. L. Moroz and W. Tan, Anal. Chem., 2008, 80, 3025 CrossRef CAS.
- X. He, K. Wang, W. Tan, B. Liu, X. Lin, C. He, D. Li, S. Huang and J. Li, J. Am. Chem. Soc., 2003, 125, 7168–7169 CrossRef CAS.
- C. Chen, Y. Liu, C. Wu, C. Yeh, M. Su and Y. Wu, Adv. Mater., 2005, 17, 404–407 CrossRef CAS.
- D. S. Seferos, D. A. Giljohann, H. D. Hill, A. E. Prigodich and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 15477–15479 CrossRef CAS.
- C. Lu, C. Zhu, J. Li, J. Liu, X. Chen and H. Yang, Chem. Commun., 2010, 46, 3116–3118 RSC.
- K. J. Luebke, R. P. Balog and H. R. Garner, Nucleic Acids Res., 2003, 31, 750–758 CrossRef CAS.
- D. P. Bratu, Methods Mol. Biol., 2006, 319, 1–14 CAS.
- Y. M. W. Janssen, Lab. Invest., 1993, 69, 261–274 CAS.
- T. Schaeffter, Prog. Drug Res., 2005, 62, 15–81 CrossRef.
- V. Ntziachristos, J. Ripoll and R. Weissleder, Opt. Lett., 2002, 27, 333–335 CrossRef.
- V. Ntziachristos, C. Bremer and R. Weissleder, Eur. J. Radiol., 2003, 13, 195–208 Search PubMed.
- R. B. Mujumdar, L. A. Ernst, S. R. Mujumdar, C. J. Lewis and A. S. Waggoner, Bioconjugate Chem., 1993, 4, 105–111 CrossRef CAS.
- E. Fluhler, V. G. Burnham and L. M. Loew, Biochemistry, 1985, 24, 5749–5755 CrossRef CAS.
- L. Song, E. J. Hennink, I. T. Young and H. J. Tanke, Biophys. J., 1995, 68, 2588–2600 CrossRef CAS.
- L. Song, C. A. G. O. Varma, J. W. Verhoeven and H. J. Tanke, Biophys. J., 1996, 70, 2959–2968 CrossRef CAS.
- X. He, J. Gao, S. Gambhir and Z. Cheng, Trends Mol. Med., 2010, 16, 574–583 CrossRef CAS.
- X. He, K. Wang and Z. Cheng, WIREs Nanomed. Nanobiotechnol., 2010, 2, 349–366 CrossRef CAS.
- J. Liu, X. Yang, X. He, K. Wang, Q. Wang, Q. Guo, H. Shi, J. Huang and X. Huo, Sci. China: Chem., 2011, 54, 1157–1176 CrossRef CAS.
- S. Sixou, F. C. Szoka, G. A. Green, B. Giusti, G. Zon and D. J. Chin, Nucleic Acids Res., 1994, 22, 662–668 CrossRef CAS.
- Q. Li, G. Luan, Q. Guo and J. Liang, Nucleic Acids Res., 2002, 30, e5 CrossRef.
- A. Tsuji, H. Koshimoto, Y. Sato, M. Hirano, Y. Sei-lida, S. Kondo and K. Ishibashi, Biophys. J., 2000, 78, 3260–3274 CrossRef CAS.
- C. J. Yang, K. Martinez, H. Lin and W. Tan, J. Am. Chem. Soc., 2006, 128, 9986–9987 CrossRef CAS.
- S. Sando and E. T. Kool, J. Am. Chem. Soc., 2002, 124, 9686–9687 CrossRef CAS.
- H. Abe and E. T. Kool, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 263–268 CrossRef CAS.
- S. Tyagi and F. R. Kramer, Nat. Biotechnol., 1996, 14, 303–308 CrossRef CAS.
- S. Tyagi, D. P. Bratu and F. R. Kramer, Nat. Biotechnol., 1998, 16, 49–53 CrossRef CAS.
- P. J. Santangelo, B. Nix, A. Tsourkas and G. Bao, Nucleic Acids Res., 2004, 32, e57 CrossRef.
- A. G. Tkachenko, H. Xie, D. Coleman, W. Glomm, J. Ryan, M. F. Anderson, S. Franzen and D. L. Feldheim, J. Am. Chem. Soc., 2003, 125, 4700–4701 CrossRef CAS.
- N. L. Rosi, D. A. Giljohann, C. S. Thaxton, A. K. R. Lytton-Jean, M. S. Han and C. A. Mirkin, Science, 2006, 312, 1027–1030 CrossRef CAS.
- C. D. Medley, J. E. Smith, Z. Tang, Y. Wu, S. Bamrungsap and W. Tan, Anal. Chem., 2008, 80, 1067–1072 CrossRef CAS.
- A. E. Prigodich, D. S. Seferos, M. D. Massich, D. A. Giljohann, B. C. Lane and C. A. Mirkin, ACS Nano, 2009, 3, 2147–2152 CrossRef CAS.
- A. Jayagopal, K. C. Halfpenny, J. W. Perez and D. W. Wright, J. Am. Chem. Soc., 2010, 132, 9789–9796 CrossRef CAS.
- S. R. Harry, D. J. Hicks, K. I. Amiri and D. W. Wright, Chem. Commun., 2010, 46, 5557–5559 RSC.
- M. Hazani, R. Naaman, F. Hennrich and M. M. Kappes, Nano Lett., 2003, 3, 153–155 CrossRef CAS.
- E. K. Hobbie, B. J. Bauer, F. Stephens, M. L. Becker, P. McGuiggan, S. D. Hudson and H. Wang, Langmuir, 2005, 21, 10284–10287 CrossRef CAS.
- J. Chlopek, B. Czajkowska, B. Szaraniec, E. Frackowiak, K. Szostak and F. Beguin, Carbon, 2006, 44, 1106–1111 CrossRef CAS.
- L. Lacerda, A. Bianco, M. Prato and K. Kostarelos, Adv. Drug Delivery Rev., 2006, 58, 1460–1470 CrossRef CAS.
- Y. Wu, J. A. Phillips, H. Liu, R. Yang and W. Tan, ACS Nano, 2008, 2, 2023–2028 CrossRef CAS.
- Z. Liu, J. T. Robinson, X. Sun and H. Dai, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef CAS.
- X. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano Res., 2008, 1, 203–212 CrossRef CAS.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS.
- H. J. Jang, Y. K. Kim, H. M. Kwon, W. S. Yeo, D. E. Kim and D. H. Min, Angew. Chem., 2010, 122, 5839–5843 CrossRef.
- H. Wang, Q. Zhang, X. Chu, T. Chen, J. Ge and R. Yu, Angew. Chem., 2011, 123, 7203–7207 CrossRef.
- C. Lu, H. Yang, C. L. Zhu, X. Chen and G. Chen, Angew. Chem., 2009, 121, 4879–4881 CrossRef.
- H. W. C. Postma, Nano Lett., 2010, 10, 420–425 CrossRef CAS.
- C. Lu, J. Li, J. Liu, H. Yang, X. Chen and G. Chen, Chem.–Eur. J., 2010, 16, 4889–4894 CrossRef CAS.
- R. M. Dirks and N. A. Pierce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15275–15278 CrossRef CAS.
- J. Huang, Y. Wu, Y. Chen, Z. Zhu, X. Yang, C. J. Yang, K. Wang and W. Tan, Angew. Chem., Int. Ed., 2011, 50, 401–404 CrossRef CAS.
- F. Wang, J. Elbaz, R. Orbach, N. Magen and I. Willner, J. Am. Chem. Soc., 2011, 133, 17149–17151 CrossRef CAS.
- S. Shimron, F. Wang, R. Orbach and I. Willner, Anal. Chem., 2012, 84, 1042–1048 CrossRef CAS.
- H. M. T. Choi, J. Y. Chang, L. A. Trinh, J. E. Padilla, S. E. Fraser and N. A. Pierce, Nat. Biotechnol., 2010, 28, 1208–1212 CrossRef CAS.
- S. Jayasena, Clin. Chem., 1999, 45, 1628–1650 CAS.
- E. N. Brody and L. Gold, Rev. Mol. Biotechnol., 2000, 74, 5–13 CrossRef CAS.
- D. Zheng, D. S. Seferos, D. A. Giljohann, P. C. Patel and C. A. Mirkin, Nano Lett., 2009, 9, 3258–3261 CrossRef CAS.
- Y. Wang, Z. Li, D. Hu, C. Lin, J. Li and Y. Lin, J. Am. Chem. Soc., 2010, 132, 9274–9276 CrossRef CAS.
- H. Chen, C. Medley, K. Sefah, D. Shangguan, Z. Tang, L. Meng, J. E. Smith and W. Tan, ChemMedChem, 2008, 3, 991–1001 CrossRef CAS.
- K. Sefah, Z. Tang, D. Shangguan and W. Tan, Leukemia, 2009, 23, 235–244 CrossRef CAS.
- Z. Zhao, L. Xu, X. Shi, W. Tan, X. Fang and D. Shangguan, Analyst, 2009, 134, 1808–1814 RSC.
- J. Herr, J. Smith, C. Medley, D. Shangguan and W. Tan, Anal. Chem., 2006, 18, 2918–2924 CrossRef.
- J. Smith, C. Medley, Z. Tang, D. Shangguan, C. Lofton and W. Tan, Anal. Chem., 2007, 79, 3075–3082 CrossRef CAS.
- W. J. Kang, J. R. Chae, Y. L. Cho, J. Lee and S. Kim, Small, 2009, 22, 2519–2522 CrossRef.
- J. Yin, X. He, K. Wang, Z. Qing, X. Wu, H. Shi and X. Yang, Nanoscale, 2012, 4, 110–112 RSC.
- R. Montesano, J. Roth, A. Robert and L. Orci, Nature, 1982, 296, 651–653 CrossRef CAS.
- C. C. Overly, K. D. Lee, E. Berthiaumet and P. J. Hollenbeck, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 3156–3160 CrossRef CAS.
- S. Modi, M. G. Swetha, D. Goswami, G. D. Gupta, S. Mayor and Y. Krishnan, Nat. Nanotechnol., 2009, 4, 325–330 CrossRef CAS.
- S. Surana, J. M. Bhat, S. P. Koushika and Y. Krishnan, Nat. Commun., 2011, 2, 340 CrossRef.
- W. Zhao, S. Schafer, J. Choi, Y. J. Yamanaka, M. L. Lombardi, S. Bose, A. L. Carlson, J. A. Phillips, W. Teo, I. A. Droujinine, C. H. Cui, R. K. Jain, J. Lammerding, J. C. Love, C. P. Lin, D. Sarkar, R. Karnik and J. M. Karp, Nat. Nanotechnol., 2011, 6, 524–531 CrossRef CAS.
- T. Tokunaga, S. Namiki, K. Yamada, T. Imaishi, H. Nonaka, K. Hirose and S. Sando, J. Am. Chem. Soc., 2012, 134, 9561–9564 CrossRef CAS.
- H. Shi, Z. Tang, Y. Kim, H. Nie, Y. Huang, X. He, K. Deng, K. Wang and W. Tan, Chem.–Asian J., 2010, 5, 2209–2213 CrossRef CAS.
- H. Shi, X. He, K. Wang, X. Wu, X. Ye, Q. Guo, W. Tan, Z. Qing, X. Yang and B. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3900–3905 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |