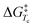Chair interconversion and reactivity of mannuronic acid esters†
Received
27th August 2013
, Accepted 10th October 2013
First published on 11th October 2013
Abstract
Mannopyranosyluronic acids display a very unusual conformation behavior in that they often prefer to adopt a 1C4 chair conformation. They are endowed with a strikingly high reactivity when used in a glycosylation reaction as a glycosyl donor. To investigate the unusual conformational behavior a series of mannuronic acid ester derivatives, comprising anomeric triflate species and O-methyl glycosides, was examined by dynamic NMR experiments, through lineshape analysis of 1H and 19F NMR spectra at various temperatures from −80 °C to 0 °C. Exchange rates between 4C1 and 1C4 chair conformations were found to depend on the electronic properties and the size of the C2 substituent (F, N3 or OBn) and the aglycon, with higher exchange rates for the glycosyl triflates and smaller C2 substituents. Low temperature 19F exchange spectroscopy experiments showed that the covalently bound anomeric triflates did not exchange with free triflate species present in the reaction mixture. To relate the conformational behavior of the intermediate triflates to their reactivity in a glycosylation reaction, their relative reactivity was determined via competition reactions monitored by 1H NMR spectroscopy at low temperature. The 2-O-benzyl ether compound was found to be most reactive whereas the 2-fluoro compound – the most flexible of the studied compounds – was least reactive. Whereas the ring-flip of the mannuronic acids is important for the enhanced reactivity of the donors, the rate of the ring-flip has little influence on the relative reactivity.
Introduction
The discovery of covalently linked anomeric triflates as reactive intermediates in a glycosylation reaction has provided new perspectives on the activation of glycosyl donors and the stereochemical course of a glycosylation reaction.1 Crich and co-workers were the first to report that glycosyl donors could be transformed into detectable anomeric triflate intermediates, which can be displaced by an incoming nucleophile to provide the glycosylation products. The displacement of the anomeric triflate can proceed through a continuum of mechanisms, ranging from a dissociative SN1-type process to an associative SN2-type reaction, depending on the reactivity of the incoming nucleophile and the stability/reactivity of the intermediate triflate. In almost all cases reported, the anomeric triflate takes up an axial orientation to benefit from the anomeric effect. For example, the seminal work of Crich and co-workers has shown that 4,6-benzylidene mannosyl donors can be transformed into axially oriented α-triflates that can be substituted with inversion of stereochemistry at the anomeric center to provide the β-linked products. In the context of the assembly of mannuronic acid alginates, which are composed of repeating (1→4)-linked β-D-mannuronic acid monomers, we studied the activation and glycosylation of a variety of mannuronic acid donors.2 In line with the 4,6-benzylidene mannosyl donor case, we found that mannuronic acid donors can be transformed into the corresponding anomeric triflates and that glycosidation of these species proceeds in a highly stereoselective manner to provide the cis-linked products. However, in sharp contrast to the vast majority of anomeric triflates reported to date, the mannuronic acid derived anomeric triflates preferentially occupy the 1C4 conformation, which places the anomeric triflate in an equatorial orientation (see Scheme 1). In addition, the carbohydrate ring-flip from the 4C1 to the 1C4 conformation leads to three sterically unfavorable axial substituents on the mannuronic acid.3,4 The conformational flexibility and the possibility to adopt the ‘inverted’ 1C4 conformation also leads to an unusually high reactivity of the mannuronic acid donors, which are significantly more reactive than what would be expected based on the presence of the electron withdrawing C5 carboxylic acid ester.5 To better understand the reasons for the unexpected ring-flip and whether the rate of ring interconversion is important for the reactivity of the mannuronic acid donors we have now studied a set of mannuronic acid triflates by dynamic NMR (DNMR) to obtain thermodynamic parameters for the ring interconversion processes.6 Notably, detailed experimental studies of the ring conformational exchange rates in carbohydrates and the associated energy barriers are quite rare,‡ because dynamic NMR studies of carbohydrates are often hampered by large population differences, high exchange rates at ambient temperature and poor solubility in organic solvents of unprotected carbohydrates, which limits the experimental temperature window.§ The flexible mannuronic acid system presents a unique opportunity to investigate how the substituents on the carbohydrate core influence the conformational behavior of the ring.
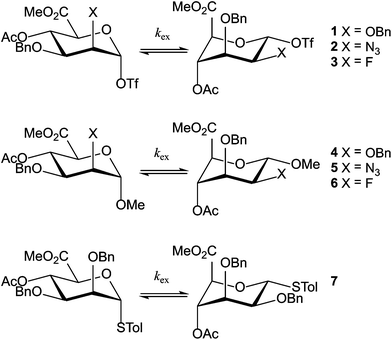 |
| | Scheme 1 Mannuronic acid esters 1–7. | |
Results
Experimental set-up
In this study the rates of the conformational transitions for a series of fully protected α-D-mannuronic acid esters are addressed to gain knowledge about how the substituents on the ring influence the conformational behavior of the mannuronic acids and what influence this conformational behavior has on the reactivity of the studied compounds. To this end we have investigated mannuronic acids, differing in the substituent at the C2 position, being either a benzyloxy, azido or fluoride moiety. We have directed our attention towards the anomeric triflates (1, 2 and 3), because these represent essential intermediates in the glycosylation reactions in which they feature, and relate the conformational properties to those of the O-methyl α-D-mannosides 4, 5 and 6, and thiomannoside 7. Triflates 1, 2 and 3 were generated from their β-thioglycoside precursors, which are conformationally stable and reside exclusively in the 4C1 conformation, through activation with diphenylsulfoxide/trifluoromethanesulfonic acid anhydride in CD2Cl2 at −80 °C (see the Experimental section for details).¶ Using dynamic NMR we determined the ratio of the two chair conformers and the interconversion rates between the conformers. With these and by NMR simulation of parts of the spectra we accurately determined the thermodynamic data for the interconversion of the two conformers. We have also been able to establish the scalar coupling J45 as a rapid and reliable indicator of the ring conformer ratio.
Dynamic NMR spectroscopy
In order to explore the conformational properties we have investigated compounds 1–7 at temperature intervals of 10 °C. Temperature arrays of 1H NMR spectra were recorded for the triflates from −80 °C to the decomposition temperature (–40 °C for benzyl ether 1, −30 °C for azide 2 and −20 °C for fluoride 3), and from −80 °C to 0 °C for methyl mannosides 4–6 and thiomannoside 7. Because the interconversion of the two chair conformers of the C2-fluoro mannuronic acids 3 and 6 was so fast, no separation of the signals was observed in the 1H NMR spectra of these compounds even at −80 °C. Therefore we switched to the use of 19F NMR spectroscopy, because the larger resonance frequency difference (Δν) of the fluorine nuclei did allow for the observation of the two conformers. As previously reported3 the spectral appearances, i.e. the lineshape and relative integrals, were retained when raising and lowering the experimental temperature.
In Fig. 1, part of the 1H NMR spectrum of mannosyl triflate 2 is depicted, showing the coalescence of the signals corresponding to H1 and H4 in the range of −80 °C to −30 °C. Coalescence of signals corresponding to other hydrogen nuclei was also observed, but monitoring the downfield H1 and H4 resonances is straightforward due to the comparatively low spectral crowding in the region of 5–6 ppm. These peaks were also used in the simulation of the spin system. From the spectra it becomes clear that the azidomannuronic acid 2 has a preference for the 1C4 conformation and a similar picture emerges for the C2-O-benzyl triflate 1 (see Table 1, entry 1). In the conformer mixture of the O-methyl mannuronic acids 4 and 5 on the other hand the 4C1 conformer prevails (Table 1, entries 4 and 5). For the thiomannoside 7 the 1C4 conformation is slightly more favored (Table 1, entry 7).
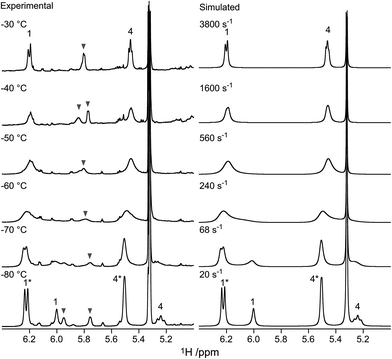 |
| | Fig. 1 Experimental and simulated NMR spectra of the H1 and H4 signals of anomeric triflate 2. Signals arising from the 1C4 conformer are marked with asterisks. The signal at 5.36 ppm originates from residual dichloromethane-d. The signals denoted with grey triangles are unknown byproducts formed upon activation. | |
Table 1 Thermodynamic data for compounds 1–7 at −70 °C
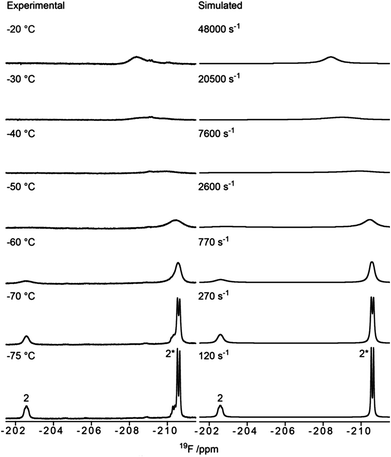
As noted above, we resorted to the use of 19F NMR to study the dynamic equilibrium between the two chair conformers of the fluoro compounds 3 and 6. The 1H NMR spectrum of 3 (recorded at −20 °C) showed a 3JH1,H2 coupling constant of 6.5 Hz, indicating a strong preference for the 1C4 conformation (>60%), since a mannopyranose in the 4C1 conformation has 3JH1,H2 < 2 Hz. Below −50 °C, the 19F NMR spectra of compound 3 showed two signals with an intensity ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (see Fig. 2, and Table 1, entry 3). Due to the preference for the 1C4 conformation of 3, the major signal with an upfield chemical shift of −210.6 ppm was assigned to the 1C4 conformation and the minor downfield-shifted signal at −202.6 ppm to the 4C1 conformation. These assignments are in agreement with those for F2 of compound 6, which exists primarily as the 4C1 conformer (see Table 1, entry 6 and ESI†). Notably, in the 19F spectrum of compound 3 recorded at −75 °C (Fig. 3) separate signals for the covalently bound triflate groups of the two conformers of compound 3 were observed between −75 and −76 ppm, flanked by signals of the triflic anhydride (downfield) and anionic triflate complexes (upfield). The latter resonances were assigned in accordance with the reports of Turnbull and co-workers7 and Crich and Sun1 and are based on peak intensity. Through the use of 2D 19F,19F-EXSY experiments we have been able to establish the covalent nature of the anomeric triflate in compound 3. At a mixing time of 100 ms cross-peaks resulting from the conformational exchange of 3 were observed for the F2 resonances (Fig. 4a) and the anomeric triflate nuclei (Fig. 4b), respectively. Cross-peaks were not observed between the glycosyl triflates and the anionic triflate species present in solution, even at longer mixing times and increased temperature (500 ms, −20 °C), indicating that there is no significant exchange of the triflate ions in solution and the covalently bound triflates.
:1 (see Fig. 2, and Table 1, entry 3). Due to the preference for the 1C4 conformation of 3, the major signal with an upfield chemical shift of −210.6 ppm was assigned to the 1C4 conformation and the minor downfield-shifted signal at −202.6 ppm to the 4C1 conformation. These assignments are in agreement with those for F2 of compound 6, which exists primarily as the 4C1 conformer (see Table 1, entry 6 and ESI†). Notably, in the 19F spectrum of compound 3 recorded at −75 °C (Fig. 3) separate signals for the covalently bound triflate groups of the two conformers of compound 3 were observed between −75 and −76 ppm, flanked by signals of the triflic anhydride (downfield) and anionic triflate complexes (upfield). The latter resonances were assigned in accordance with the reports of Turnbull and co-workers7 and Crich and Sun1 and are based on peak intensity. Through the use of 2D 19F,19F-EXSY experiments we have been able to establish the covalent nature of the anomeric triflate in compound 3. At a mixing time of 100 ms cross-peaks resulting from the conformational exchange of 3 were observed for the F2 resonances (Fig. 4a) and the anomeric triflate nuclei (Fig. 4b), respectively. Cross-peaks were not observed between the glycosyl triflates and the anionic triflate species present in solution, even at longer mixing times and increased temperature (500 ms, −20 °C), indicating that there is no significant exchange of the triflate ions in solution and the covalently bound triflates.
 |
| | Fig. 2 Experimental and simulated 19F NMR spectra of anomeric triflate 3. The signal arising from F2 of the 1C4 conformer is marked with an asterisk (*). | |
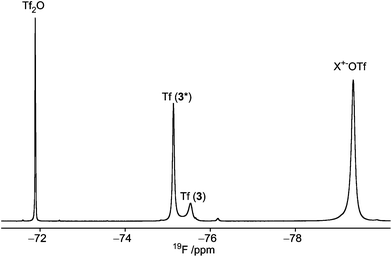 |
| | Fig. 3
19F NMR spectrum of 3, at −75 °C, in which the signal arising from the 1C4 conformer is marked with an asterisk (*). The relative ratio χ = 1:3 between 3 and 3*. X+ = Ph2SOTf+, (Ph2S)2O2+ according to Turnbull and co-workers.7 | |
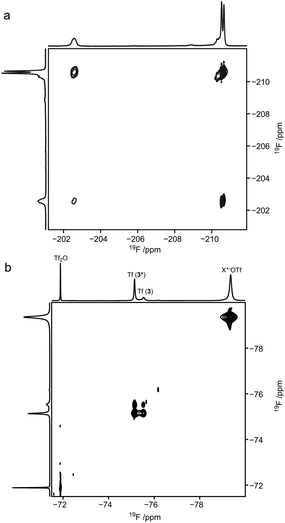 |
| | Fig. 4 2D 19F,19F-EXSY-spectrum of (a) the upfield region showing the F2 resonances of compound 3 and (b) the downfield region showing the triflate species, at −70 °C; mixing time 0.1 s. | |
Dynamics
We next established the conformational exchange rates (kex = k1 + k−1, Scheme 1) of compounds 1–7 by NMR spin simulation using the experimentally acquired spectra by iterative fitting of the line widths, intensities and exchange rates (Fig. 1 and 2, and S1–S5†). The lineshape of the NMR spectra are strongly correlated to the lifetimes of the resonances at different energy levels. Short life times, i.e. high exchange rates, yield coalesced spectra, while slow exchange gives separated resonances. In the intermediate exchange rate, where the exchange rate is of the same magnitude as the frequency difference between the resonances, broadening of the signals occurs as a result of the Heisenberg uncertainty principle with respect to the energy levels, i.e., ΔE = hΔν1/2, where Δν1/2 equals the full width at half maximum of the resonance line.6 Spin simulation enables quantification of the variables defining the lineshape. For compounds 1, 2, 5, and 71H NMR spectra were simulated, using the signals of H1 and H4. For compound 4, in which H1 was obstructed by severe spectral overlap, only the H4 resonance was utilized. For the 2-fluoro compounds 3 and 619F NMR spectra were used. The determined exchange rates are listed in Table 2, from which it immediately becomes clear that the interconversion of the two chair conformers is significantly faster for the anomeric triflates than the corresponding methyl glycosides. The C2–F triflate 3 flips the fastest of the examined set of compounds. The energy required to flip the mannuronic acid ring, ΔG‡, was calculated via a redistributed version of the Eyring equation:| | 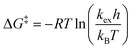 | (1) |
where R is the gas constant, h is Planck's constant and kB is Boltzmann's constant. The ΔG‡ for compounds 1–7 at −70 °C are listed in Table 1. From this table it is clear that the ΔG‡ for the ring flip of methyl glycosides 4–6 was equal, within the error margin, to ∼46 kJ mol−1, whereas the energy barriers were significantly lower for the glycosyl triflates 1–3, with fluoride 3 requiring the least energy for its conformational change (ΔG‡3 = 39.6 kJ mol−1). The highest energy barrier was observed for thioglycoside 7: 48.4 kJ mol−1. To verify the ΔG‡ values obtained from the simulations, a simpler calculation of the transition state energy barriers (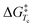 ) was performed based on the coalescence temperature (Tc) and the maximum frequency difference (Δν):6
) was performed based on the coalescence temperature (Tc) and the maximum frequency difference (Δν):6| | 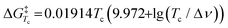 | (2) |
Table 2 Exchange rates (s−1) of compounds 1–7 at different temperatures
|
T (°C) |
1
|
2
|
3
|
4
|
5
|
6
|
7
|
| 0 |
|
|
|
1600 |
10000 |
6500 |
1400 |
| −20 |
|
|
48000 |
330 |
1824 |
970 |
290 |
| −30 |
|
3800 |
20500 |
180 |
640 |
291 |
|
| −40 |
500 |
1600 |
7600 |
71 |
240 |
103 |
42 |
| −50 |
160 |
580 |
2600 |
23 |
68 |
32 |
|
| −60 |
77 |
240 |
770 |
10 |
21 |
14 |
6.0 |
| −70 |
23 |
68 |
270 |
5.0 |
5.2 |
4.7 |
|
| −75 |
|
|
120 |
|
|
3.7 |
|
| −80 |
6.4 |
20 |
|
1.3 |
1.8 |
|
1.0 |
The resulting energy barriers, also tabulated in Table 1, are similar to those obtained from the DNMR-simulations and the observed trend is retained, giving credibility to the deployed simulations.
Conformation and J couplings
To allow for rapid determination of the ratio of the two chair conformers of the mannuronic acid esters, we analyzed the JHH coupling constants from the coalesced spectra of compounds 3–7. These were extracted from the 1D 1H NMR spectra using NMR spin simulation,8 and are summarized in Table 3. The signals in the coalesced spectra of compounds 1 and 2 were too broad to enable coupling constant analysis, and were therefore omitted. The JHH coupling constants of the compounds were plotted against the percentage of the 4C1 conformation in the equilibrium populations, as determined by integration of the separate peaks at the respective lowest experimental temperatures in 1H or 19F NMR spectra, resulting in the graph displayed in Fig. 5. Several coupling constants display linear correlations to the population distribution, but with different slopes for different couplings, depending on the position of the protons on the ring, and the substituent attached. From the data in Fig. 5 the J45 coupling constant emerges as the best reporter on the ring conformational distribution of this class of compounds: it has a high correlation coefficient (R2 = 0.976) and the largest coupling constant amplitude. Also the J12 coupling constant can be of use in complementing J45 in a conformational analysis of this kind. However, due to substituent changes at C1 and C2 these coupling constants may be less useful. From the linear fit of the J45 coupling constants against the population of the 4C1 conformation eqn (3) is proposed as a good estimate of the populations of an α-mannuronic acid ester in rapid equilibrium between the 4C1 and 1C4 conformations:||| | | P(4C1) = 0.142 × J45 − 0.311 | (3) |
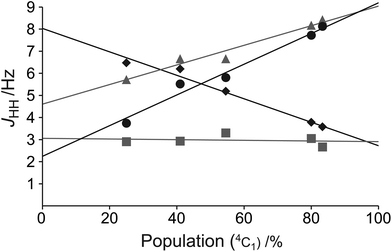 |
| | Fig. 5 The coupling constants J12 (black diamonds), J23 (grey squares), J34 (grey triangles), and J45 (black circles) of compounds 3–7 as a function of conformational distribution. | |
Table 3 Population of chair conformers (%) and coupling constants (Hz) in coalesced spectra of compounds 3–7 at 0 °C and of the separate conformers and weighted average (![[X with combining macron]](https://www.rsc.org/images/entities/i_char_0058_0304.gif) ) of compounds 5 and 7 at −80 °C
) of compounds 5 and 7 at −80 °C
| # |
4C1 |
1C4 |
J
12
|
J
23
|
J
34
|
J
45
|
|
At −20 °C.
|
|
3
|
25 |
75 |
6.473 |
2.902 |
5.716 |
3.737 |
|
4
|
80 |
20 |
3.784 |
3.052 |
8.172 |
7.721 |
|
5
|
55 |
45 |
5.187 |
3.302 |
6.657 |
5.807 |
|
6
|
83 |
17 |
3.577 |
2.665 |
8.425 |
8.126 |
|
7
|
41 |
59 |
6.197 |
2.930 |
6.656 |
5.515 |
|
5
|
100 |
0 |
1.664 |
3.525 |
9.696 |
10.202 |
|
5
|
0 |
100 |
8.820 |
2.777 |
3.620 |
1.508 |
![[5 with combining macron]](https://www.rsc.org/images/entities/char_0035_0304.gif)
|
55 |
45 |
4.917 |
3.185 |
6.934 |
6.250 |
|
7
|
100 |
0 |
1.536 |
2.944 |
10.091 |
10.065 |
|
7
|
0 |
100 |
10.236 |
2.126 |
4.074 |
1.436 |
![[7 with combining macron]](https://www.rsc.org/images/entities/char_0037_0304.gif)
|
41 |
59 |
6.669 |
2.461 |
6.541 |
4.974 |
For compounds 5 and 7 the signals of the entire spin systems of the two respective chair conformers were well resolved which allowed J coupling analysis by spin simulation (Table 3). The weighted average of the determined coupling constants, with respect to populations, was in good agreement (ΔJ ≤ 0.5 Hz) with the couplings measured for the coalesced spectra, which indicates that the equilibrium populations of compounds 5 and 7 are not largely affected by temperature and illustrate the accuracy of the methods deployed for spectral analysis. The application of empirical J coupling data for conformational analysis of coalesced spectra is a good complement to the methodology based on Karplus-type relationships, employed in other studies.9,10
Discussion
Tables 1 and 2 summarize all data we obtained for the dynamic conformational equilibria of compounds 1–7. It is clear from Table 1 that the anomeric triflates and methyl mannosides have a different conformational preference. Whereas the 1C4 conformation dominates the equilibrium of the glycosyl triflates 1–3, the 4C1 conformation is favored for the methyl glycosides 4–6. For the triflates the 1C4 conformer becomes more important for the smaller and more electronegative substituents. Generally, α-D-mannopyranosides adopt a 4C1 conformation, because this conformation places most ring substituents in an equatorial arrangement thereby minimizing steric strain. In addition, the axially oriented C1-substituents benefit from a stabilizing anomeric effect in this constellation. We have previously postulated that the preference of the mannuronic acid triflates to adopt the counterintuitive 1C4 conformation could be explained by assuming that the anomeric center in these molecules bears a significant amount of positive charge.4 To accommodate this positive charge best, the structures take up a conformation which allows for the most stabilization (the least destabilization) of the positive charge, which is the ‘axially rich’ 1C4 conformation.11 The data presented here support this hypothesis. The C5 carboxylic acid ester plays a decisive role since it withdraws electron density from the anomeric center, thereby adding to the carbocation character of this center. At the same time it can stabilize the partial positive charge if it is positioned properly in space, i.e., in an axial orientation in the 1C4 conformation. A further indication of the interaction of the C5 carboxylic acid ester is found in the chemical shift of the anomeric proton in both chair conformers. Generally, an equatorial anomeric proton is expected to resonate downfield to an axial one, with a chemical sift difference (δppm) of 0.5–0.7 ppm.12 However, for compounds 1, 2, 4 and 5 the signals corresponding to the axial H1 (1C4 conformation) appear downfield with respect to the equatorial H1 (4C1 conformation) (δppm = 0.2–0.4 ppm), corresponding to a downfield shift displacement of roughly 1 ppm. This large chemical shift change may hypothetically be explained by a through space interaction of the anomeric hydrogen atom and the carbonyl oxygen, as depicted in Fig. 6.13,14 For thioglycoside 7 the downfield shift of the 1C4 H1 signal is smaller, ∼0.1 ppm, which can be attributed to the reduced electronegativity of the thiol aglycone with respect to the oxygen substituents in 1–6 and thereby a lower hydrogen bond accepting ability of H1.
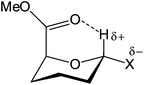 |
| | Fig. 6 Proposed non-classical intramolecular hydrogen bond. | |
From Table 1, two trends emerge for the exchange rates of compounds 1–7: (1) The exchange rate increases with increased electronegativity of the aglycone (i.e. kOTf > kOMe > kSTol) and (2) a smaller substituent at C2 leads to a faster ring inversion for the glycosyl triflates 1–3 (i.e. kF > kN3 > kOBn). The first trend can be rationalized by the increased oxocarbenium character at C1 in the compounds with a more electronegative aglycone. This leads to an increased C1–aglycone bond length and a flattening of the ring. It can be envisaged that the 4C1–1C4 interconversion proceeds through a pseudorotational itinerary via a 3H4-like transition state, a structure in which all ring substituents are positioned such that they allow for stabilization of the partial positive charge at the anomeric center.2 It can also be argued that the size of the aglycone is at the basis of the observed trend, with the largest aglycone, the thiotolyl group, flipping the slowest. The longer C1–OTf bond leads to a decrease in steric interaction of this substituent relative to the OMe aglycone, accounting for the faster flipping of compound 1 with respect to compound 4. The second trend revealed in Table 1 can be rationalized by steric interactions, in combination with the electronic factor outlined above. The compound with the smallest, most electron withdrawing C2 substituent shows the fastest ring interconversion.
Glycosylation reactions
With the conformational properties of compounds 1–3 established, we investigated whether an increased conformational exchange rate corresponds to an increased tendency to react as an electrophile in a glycosylation reaction. To this end we performed a series of competition glycosylation reactions, which were followed by 1H NMR spectroscopy. Thus, a 1:1 mixture of glycosyl triflates 2 and 3 was generated from the 1:1 mixture of parent thioglycosides using Ph2SO–Tf2O in CD2Cl2 at −80 °C (Fig. 7). Subsequently methanol was added in portion sizes of 0.5 molar equivalents. Rapid consumption of the 2-azido compound 2 was observed in the sequentially recorded 1H NMR spectra, while the 2-fluoro compound 3 was virtually unaffected even after addition of 2.5 equivalents of methanol. Upon addition of an excess of methanol and an increase of the sample temperature, complete consumption of 3 was observed. The resulting spectra displayed a complex mixture, mainly consisting of anomeric mixtures of methyl glycosides. In an analogous competition experiment between the 2-O-Bn compound 1 and the 2-azido compound 2, consumption of donor 1 was slightly faster than that of 2, although the difference in reactivity was not as significant as observed for the C2-azido and C2-fluoro compounds (Fig. S6†). Clearly, the trend of reaction rates is the opposite of that observed for the conformational exchange, namely 1 ≥ 2 > 3, and therefore we conclude that enhanced ring flipping does not add significantly to the reactivity of the system. The glycosylation reactivity of the triflate intermediates does follow the same trend observed for the reactivity of the thioglycoside donors, as we had previously established. The relative reactivities are also in line with the decomposition temperatures of the triflates, which are 40 °C, −30 °C and −20 °C for 1, 2 and 3, respectively. Thus, the electronegativity of the substituent at C2 is the most decisive factor in determining the reactivity of the mannuronate acid esters.
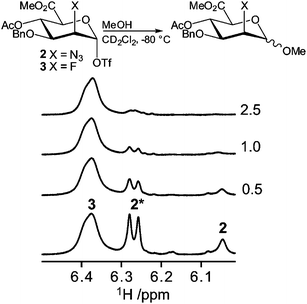 |
| | Fig. 7 Schematic description of the reactions (top) and anomeric region of the 1H NMR spectra (bottom) from the competition experiment of anomeric triflates 2 and 3. The numbers in the right margin denote the number of methanol equivalents added. The signal arising from the 1C4 conformer is marked with an asterisk. | |
Conclusions
Mannuronic acid donors can be transformed into anomeric triflates upon activation prior to or during a glycosylation reaction. Besides the excellent β-stereoselectivity displayed by the mannuronic acid esters in a glycosylation reaction, these donors stand out because of their high reactivity and uncommon conformational behavior. Depending on the exact substitution pattern, α-mannuronic acid esters preferentially populate the common 4C1-chair conformation or the unusual 1C4-chair conformation. The latter conformation is preferred for the mannuronic acid ester α-triflates, which is rather unusual given the absence of a stabilizing anomeric effect and the presence of three axial substituents in this constellation. The present work has focused on studying the conformational behavior and reactivity of three mannuronic acid triflates, bearing different substituents at C2, i.e. a benzyloxy group, an azide and a fluoride, using dynamic NMR spectroscopy and simulation techniques. Because conditions that allow for the study of conformational exchange rates are rarely encountered for fully protected carbohydrates, the mannuronic acid system at hand has provided a unique opportunity to perform these studies. The equilibrium of the 4C1- and 1C4-chair conformers and the exchange rate between them depend on the substituents at both the C2 position and the anomeric center. The studied anomeric triflates preferentially adopt a 1C4-chair conformation, and this preference is stronger for the mannuronic acids having a stronger electron withdrawing group appended at C2, indicating that the generation of positive charge at the anomeric center15 is an important driving force for the ring-flip. The exchange rate for the ring-flip is also significantly affected by the C2 substituent and the mannuronic acids interconvert faster when a small, strongly electron withdrawing substituent is present. The anomeric triflates were shown to flip faster than the corresponding methyl mannosides and mannuronic acid with a thiotolyl aglycone. The fact that the anomeric triflates bear a significant positive charge at their anomeric centers and therefore are inclined to flatten their chair structure to accommodate this charge can account for the relatively high rate of ring interconversion of the anomeric triflates. Competition glycosylation experiments, conducted in an NMR tube to closely follow the disappearance of the mannuronic acid donors, have shown that the enhanced ring-flipping rate of the mannuronic acids with a stronger electron withdrawing substituent at C2 does not significantly add to the reactivity of the system. The electron withdrawing effect of the C2 substituent seems to be a more important factor in determining the reactivity. Thus, whereas the ring-flip itself is important to account for the enhanced reactivity of the mannuronic acids, the rate of the ring-flip has little influence.
Experimental details
General methods
NMR experiments for conformational analysis were carried out on a Bruker DMX 400 MHz spectrometer equipped with a BBI Z-gradient probe for 1H NMR measurements and a SEL probe for 19F NMR measurements. The spectrometer temperature was calibrated by monitoring the chemical shift difference of the hydroxyl and methyl protons of neat methanol.16 The ring protons (H1–H5) of the coalesced and separate spectra of compounds 1–7 were assigned by using 2D 1H,1H-COSY and 1H,13C-HSQC experiments. The 2D 19F,19F-EXSY spectra of compound 3 were acquired with a mixing time of 0.1 s and a total relaxation delay of 2.1 s. nJHH and nJHF coupling constants of compounds 3–7 were determined with the aid of the PERCH NMR spin simulation software (PERCH Solutions Ltd, Kuopio, Finland). Chemical shifts and coupling constants were altered iteratively until the simulated and experimental spectra appeared to be highly similar according to visual inspection and the total root-mean-square value was close to or below 0.1%.
Spin simulation was performed with the DNMR software featured in Topspin 2.1. The chemical shifts and coupling constants of the peaks of the respective conformational states and the equilibrium ratio were set at the lowest experimental temperature. The line-broadening parameter was fitted to the intrinsic solvent peak at each temperature. The exchange rate parameter was iteratively altered until the simulated and experimental spectra appeared to be highly similar according to visual inspection. The acquired simulated spectrum was then used as a starting point for the simulation at the next temperature level.
General procedure for low temperature NMR experiments of glycosyl triflates
The respective thioglycoside donor, 0.03 mmol, and diphenyl sulfoxide, 0.039 mmol, were co-evaporated twice from toluene. The mixture was dissolved in 0.6 mL of CD2Cl2 and transferred to an NMR-tube under an argon atmosphere. The sample mixture was inserted into the NMR spectrometer, and the system was tuned, matched, locked and shimmed at −80 °C (−75 °C for the 19F NMR experiments of compound 3). In an acetone bath (−80 °C) triflic anhydride, 0.039 mmol (7 μL), was added to the sample mixture and the tube was shaken and transferred back to the spectrometer; shimming was performed again. 1D 1H NMR and/or 19F NMR spectra were recorded subsequently whereafter the temperature was raised in steps of 10 °C.
General procedure for competition experiments by NMR
A mixture of the respective thioglycoside donors, 0.015 mmol each, and diphenyl sulfoxide, 0.033 mmol (6.7 mg), was co-evaporated from toluene twice. The mixture was dissolved in 0.6 mL of CD2Cl2 and transferred to an NMR-tube under an argon atmosphere. The sample was inserted into the NMR spectrometer, and the system was tuned, matched, locked and shimmed at −80 °C. In an acetone bath (−80 °C) triflic anhydride, 0.039 mmol (7 μL), was added to the sample mixture and the tube was shaken and transferred back to the spectrometer with subsequent shimming. Methanol was added from a stock solution prepared from 0.6 mL CD2Cl2 and 25 μL MeOH, from which portions of 7.5 μL, containing 0.0075 mmol MeOH, were added to the reaction mixture. 1H NMR spectra were recorded subsequent to each such addition. When five portions had been added, neat MeOH (2 μL) was added and another spectrum was acquired. The sample was then kept at room temperature for 0.5–3 h whereafter a final 1H NMR spectrum was recorded.
Acknowledgements
This work was supported by grants from the Swedish Research Council, the Knut and Alice Wallenberg Foundation and Stiftelsen Längmanska kulturfonden.
References
- D. Crich and S. Sun, J. Am. Chem. Soc., 1997, 119, 11217–11223 CrossRef CAS.
- M. T. C. Walvoort, G. A. van der Marel, H. S. Overkleeft and J. D. C. Codée, Chem. Sci., 2013, 4, 897–906 RSC.
- M. T. C. Walvoort, G. Lodder, J. Mazurek, H. S. Overkleeft, J. D. C. Codée and G. A. van der Marel, J. Am. Chem. Soc., 2009, 131, 12080–12081 CrossRef CAS PubMed.
- M. T. C. Walvoort, J. Dinkelaar, L. J. van den Bos, G. Lodder, H. S. Overkleeft, J. D. C. Codée and G. A. van der Marel, Carbohydr. Res., 2010, 345, 1252–1263 CrossRef CAS PubMed.
- M. T. C. Walvoort, W. de Witte, J. van Dijk, J. Dinkelaar, G. Lodder, H. S. Overkleeft, J. D. C. Codée and G. A. van der Marel, Org. Lett., 2011, 13, 4360–4363 CrossRef CAS PubMed.
-
J. Sandström, Dynamic NMR Spectroscopy, Academic Press, New York, 1982 Search PubMed.
- M. A. Fascione, S. J. Adshead, P. K. Mandal, C. A. Kilner, A. G. Leach and W. B. Turnbull, Chem.–Eur. J., 2012, 18, 2987–2997 CrossRef CAS PubMed.
- R. Laatikainen, M. Niemitz, U. Weber, J. Sundelin, T. Hassinen and J. Vepsäläinen, J. Magn. Reson., Ser. A, 1996, 120, 1–10 CrossRef CAS.
- J. Rönnols, A. Burkhardt, I. Cumpstey and G. Widmalm, Eur. J. Org. Chem., 2012, 74–84 CrossRef.
- A. Siegbahn, U. Aili, A. Ochocinska, M. Olofsson, J. Rönnols, K. Mani, G. Widmalm and U. Ellervik, Bioorg. Med. Chem., 2011, 19, 4114–4126 CrossRef CAS PubMed.
- S. Chamberland, J. W. Ziller and K. A. Woerpel, J. Am. Chem. Soc., 2005, 127, 5322–5323 CrossRef CAS PubMed.
- M. Lundborg and G. Widmalm, Anal. Chem., 2011, 83, 1514–1517 CrossRef CAS PubMed.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1619–1636 CrossRef CAS PubMed.
- M. Zierke, M. Smiesko, S. Rabbani, T. Aeschbacher, B. Cutting, F. H.-T. Allain, M. Schubert and B. Ernst, J. Am. Chem. Soc., 2013, 135, 13464–13472 CrossRef CAS PubMed.
- H. Satoh, H. S. Hansen, S. Manabe, W. F. van Gunsteren and P. H. Hünenberger, J. Chem. Theor. Comput., 2010, 6, 1783–1797 CrossRef CAS.
- C. Amman, P. Meier and A. E. Merbach, J. Magn. Reson., 1982, 46, 319–321 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Dynamic 1H and 19F NMR spectra and 1H NMR spectra from a competition experiment. See DOI: 10.1039/c3ob41747f |
| ‡ In a study from 1969 it was reported that β-D-ribopyranose tetraacetate had an inversion rate from the 4C1 to 1C4 conformation of 130 s–1 at −60 °C, equivalent to ΔG‡ = 41.5 kJ mol−1. [P. L. Durette and D. Horton, Carbohydr. Res., 1969, 10, 565–577.] |
| § Substituted cyclohexanes and piperidines are less limited by these problems, and have been studied more frequently. [G. Gill, D. M. Pawar and E. A. Noe, J. Org. Chem., 2005, 70, 10726–10731; A. D. Bain, M. Baron, S. K. Burger, V. J. Kowalewski and M. B. Belén Rodríguez, J. Phys. Chem. A, 2011, 115, 9207–9216; E. L. Eliel, D. M. Kandasamy, C. Yen and K. D. Hargrave, J. Am. Chem. Soc., 1980, 102, 3698–3707.] |
| ¶ Upon the generation of compound 1 a significant amount of an unidentified by-product was observed in the 1H NMR spectra (Fig. S1†). The by-product did not obstruct the lineshape analysis of compound 1. |
| || Applying this equation to the previously reported spectral data for methyl (phenyl 4-O-acetyl-2,3-diazido-2,3-dideoxy-1-thio-α-D-mannopyranosyl uronate) [M. T. C. Walvoort, G. Moggré, G. Lodder, H. S. Overkleeft, J. D. C. Codée and G. A. van der Marel, J. Org. Chem., 2011, 76, 7301–7315.], in which J45 = 6.5 Hz, indicates an equilibrium consisting of 61% 4C1 and 39% 1C4. |
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a