
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSI-PET-RAFT in flow: improved control over polymer brush growth†
Andriy R.
Kuzmyn
 *a,
Martijn
van Galen
abc,
Barend
van Lagen
a and
Han
Zuilhof
*ad
*a,
Martijn
van Galen
abc,
Barend
van Lagen
a and
Han
Zuilhof
*ad
aLaboratory of Organic Chemistry, Wageningen University & Research, Stippeneng 4, 6708 WE Wageningen, The Netherlands. E-mail: andrii.kuzmyn@wur.nl; han.zuilhof@wur.nl
bPhysical Chemistry and Soft Matter, Wageningen University & Research, Stippeneng 4, 6708 WE Wageningen, The Netherlands
cLaboratory of Biochemistry, Wageningen University and Research, Stippeneng 4, 6708 WE, Wageningen, the Netherlands
dSchool of Pharmaceutical Sciences and Technology, Tianjin University, 92 Weijin Road, Tianjin, 300072, China
First published on 12th June 2023
Abstract
Surface-initiated photoinduced electron transfer-reversible addition–fragmentation chain transfer (SI-PET-RAFT) provides a light-dependent tool to synthesize polymer brushes on different surfaces that tolerates oxygen and water, and does not require a metal catalyst. Here we introduce improved control over SI-PET-RAFT polymerizations via continuous flow conditions. We confirm the composition and topological structure of the brushes by X-ray photoelectron spectroscopy, ellipsometry, and AFM. The improved control compared to no-flow conditions provides prolonged linear growth of the polymer brush (up to 250 nm, where no-flow polymerization maxed out <50 nm), and improved polymerization control of the polymer brush that allows the construction of diblock polymer brushes. We further show the linear correlation between the molecular weight of the polymer brush and its dry thickness by combining ellipsometry and single-molecule force spectroscopy.
Introduction
Controlled radical polymerization (CRP), often also known as reversible deactivation radical polymerization (RDRP) techniques,1,2 provide unique means to tune the molecular weight, dispersity, block sequence, and molecular architecture of synthetic polymers.3–5 The introduction of CRPs, along with many other unique applications, also allowed the controlled formation of polymer brushes.6,7 Polymer brushes are characterized by macromolecules being bound in high surface density to a surface by a chain end or ends.6,8–11 This characteristic high surface density of polymer brushes often creates unique properties, such as preventing non-specific adsorption of proteins, other polymers, or even larger entities.12 Polymer brushes have thus been used to make surfaces antifouling,13–20 antibacterial,21 antiviral,22 bioactive,17 biointeractive,19,23,24 biomimetic,25 and/or lubricating.26,27 Those and many other applications make polymer brush-based coatings uniquely positioned for the development of widely different fields of application, including improved biosensors, tissue engineering, and biointeractive devices.13,18,20Surface-initiated atom transfer radical polymerization (SI-ATRP)13,17 and related variations of RDRP, such as supplemental activator and reducing agent (SARA),28 single-electron transfer-living radical polymerizations (SET-LRP),29 and light-triggered living radical polymerization (LT-LRP),19 are among the most common techniques to make polymer brushes, and with significant success.7 However, frequently encountered disadvantages of those techniques include the use of heavy metals and the need for an oxygen-free environment during synthesis. Those aspects hamper further bioapplications and scaling-up. The reversible addition–fragmentation transfer polymerization (RAFT),30 which typically uses a thiocarbonylthio moiety, is one the most versatile and widely used methods for polymer synthesis. In addition, the technique does not require heavy metals and can be applied to create polymer brushes via immobilization of the RAFT agent on the surface.31
The next step in the evolution of surface-initiated CRPs (SI-CRP) was the introduction of light as a reaction-inducing trigger. In RAFT-based techniques, this invoked the use of photoinduced electron transfer-reversible addition–fragmentation chain transfer (PET-RAFT).32–35 We and others further developed this method to be applied to surfaces, labelled surface-initiated PET-RAFT, or SI-PET-RAFT, to create polymer brushes with increasing levels of functionality.16,18,20,35 This technique requires only simple and mild conditions. For example, we showed it can proceed in an aqueous solution containing an edible photocatalyst,16,18,20,22 and allows for the visible-light-induced patterned growth of complex polymer brushes in a controlled fashion in the presence of oxygen in the water.16,18,20,22
A very recent development in CRPs techniques, particularly RAFT, is the introduction of flow chemistry.36–38 The main benefits of applying flow chemistry in polymer synthesis include improved synthetic precision, improved performance in photopolymerization reactions. In view of our overall goal to generate with increased precision polymer brushes for bioactive surfaces,19,39 we were thus interested to see whether also in-flow variations of SI-PET-RAFT could be developed.
The current paper aims for precisely that: the combination of the SI-PET-RAFT technique with flow polymerization, in order to introduce better control to the polymerization. To this aim, we investigated and compared the properties of a series of polymer brushes obtained by SI-PET-RAFT in continuous flow and no-flow conditions, using X-ray photoelectron spectroscopy (XPS), ellipsometry, and single-molecule force spectroscopy (SMFS). In addition, we compared the polymerization in flow and non-flow conditions in the presence of four commercially available metal-free, non-toxic xanthene-based dyes (eosin Y (EY), erythrosin B (EB), phloxine B (Ph), and rose bengal (R) (additionally see ESI Fig. S1†). We further outlined that the SI-PET-RAFT in flow allows for well-controlled polymerization conditions by reinitiation and creation of diblock patterned surface architectures.
Results and discussion
The setup for conducting the SI-PET-RAFT reactions in continuous flow and no-flow conditions was the same in the first three steps (Fig. 1additionally see ESI Fig. S2†). We start with bare silicon surfaces (∼1 × 1 cm), that were first oxidized using an air plasma for 5 min and subsequently coated with (3-aminopropyl)triethoxysilane (APTES). In the next step, APTES-functionalized surfaces were reacted with 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid N-succinimidyl ester (RAFT-NHS), yielding a RAFT agent-functionalized monolayer. Previously, we investigated the yield of this reaction to be 29 ± 4%.20 The last step of the polymerization in no-flow conditions was conducted by submerging the RAFT agent-functionalized surface in an aqueous polymerization solution. The solution contained the photocatalyst, triethanolamine (TEOA) and monomer poly(ethylene glycol) methyl ether methacrylate (MeOEGMA; Mn = 300), in molar ratios of 0.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:200. The polymerization started by irradiation of the sample with a blue LED light (410 nm) for different time periods. Under flow conditions, we placed the RAFT-functionalized surface in a flow chamber (Fig. 1b and c), and pumped the polymerization solution over the surface, with a flow rate of 30 μL min−1. The thickness of the polymerization solution on top of the surfaces was 2 mm in both no-flow and flow conditions. The polymerization was stopped by stopping the irradiation and copiously washing the surfaces with water and ethanol.
:1:200. The polymerization started by irradiation of the sample with a blue LED light (410 nm) for different time periods. Under flow conditions, we placed the RAFT-functionalized surface in a flow chamber (Fig. 1b and c), and pumped the polymerization solution over the surface, with a flow rate of 30 μL min−1. The thickness of the polymerization solution on top of the surfaces was 2 mm in both no-flow and flow conditions. The polymerization was stopped by stopping the irradiation and copiously washing the surfaces with water and ethanol.
 | ||
| Fig. 1 General setup of SI-PET-RAFT in continuous flow conditions. (a) Chemical reactions scheme of SI-PET-RAFT technique (b) photo of flow reactor (c) schematic depictions of SI-PET-RAFT in flow conditions. | ||
Fig. 2a shows a representative XPS wide-scan spectrum of a poly(MeOEGMA) layer (47 nm thickness, as measured by ellipsometry) synthesized using EY as photocatalyst in flow conditions after 60 min polymerization time. The spectra showed two main peaks for O 1s (532 eV) and C 1s (285 eV). The average ratio between O 1s:C 1s XPS peaks under flow and no-flow conditions using different xanthene photocatalysts was always 1.00:2.65 ± 0.01 (see ESI for additional wide XPS spectra of poly(MEOGMA) Tables S2–S9 and exact peaks ratios Tables S10–S17†). This ratio agrees with previously reported XPS spectra for this polymer.17,18,20 The representative XPS C 1s narrow-scan spectrum is shown in Fig. 2b (see ESI for additional C 1s XPS spectra Tables S18–S25†) and demonstrates three main peaks of carbon atoms: [C–C/H](285.0 eV):[C–O](286.5 eV):[O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O] (289.0 eV). The average ratio between [C–C/H]:[C–O]:[O–CO] peaks obtained under different polymerization conditions and in the presence of various photocatalysts was determined to be 2.8 ± 0.1:9.8 ± 0.1:1.0 (Fig. 2b). These spectra agree with a previously simulated spectrum, giving a ratio between [C–C/H]:[C O]:[O CO] of 3:10:1, reported by us.20
O] (289.0 eV). The average ratio between [C–C/H]:[C–O]:[O–CO] peaks obtained under different polymerization conditions and in the presence of various photocatalysts was determined to be 2.8 ± 0.1:9.8 ± 0.1:1.0 (Fig. 2b). These spectra agree with a previously simulated spectrum, giving a ratio between [C–C/H]:[C O]:[O CO] of 3:10:1, reported by us.20
 | ||
| Fig. 2 XPS characterization of poly(MeOEGMA) brushes obtained by SI-PET-RAFT in flow condition (polymerization time: 60 min; thickness 47 nm): representative (a) wide-scan spectrum, and (b) narrow-scan C 1s spectrum. | ||
The kinetics of the polymer brush growth in flow and no-flow conditions monomers were followed by measuring the polymer brush layer thicknesses with scanning ellipsometry (Fig. 3; see also ESI, Tables S26–S33†). The SI-PET-RAFT brush growth performed in no-flow conditions plateaus after 90–120 min at an average of 36 nm. Previously, our group18,20 and those of Boyer and Pester35 observed plateauing in the kinetics after some period of linear growth in SI-PET-RAFT polymerizations. In contrast, the polymerization in flow conditions showed linear growth without levelling off, and, consequently good control of the polymerization. We and others previously suggested that under no-flow conditions refreshing the solution of polymerization or in the presence of an oxygen-consuming agent or oxygen-free environment allowed to improve the polymerization control.18,20,35 We originally hypothesized that decreased photocatalyst activity during polymerization was one of the main reasons for the loss of polymerization control and plateauing of the kinetics. The polymerization solutions typically change color during non-flow polymerization from a shade of pink to yellow or colorless, and in fact, the slow degradation of xanthene-based dyes under similar light conditions by dehalogenation has been reported.40,41 However, as kindly suggested by a reviewer, repeated injection of fresh catalyst to the solution in no-flow conditions only marginally improved polymerization control (growing from 0 to 26% improvement over 7 h; Fig. S3†). The improvement observed under flow conditions, is thus largely related to the continuous refreshment of the polymerization solution, rather than that of the photocatalyst, although the latter has some (minor) effect.
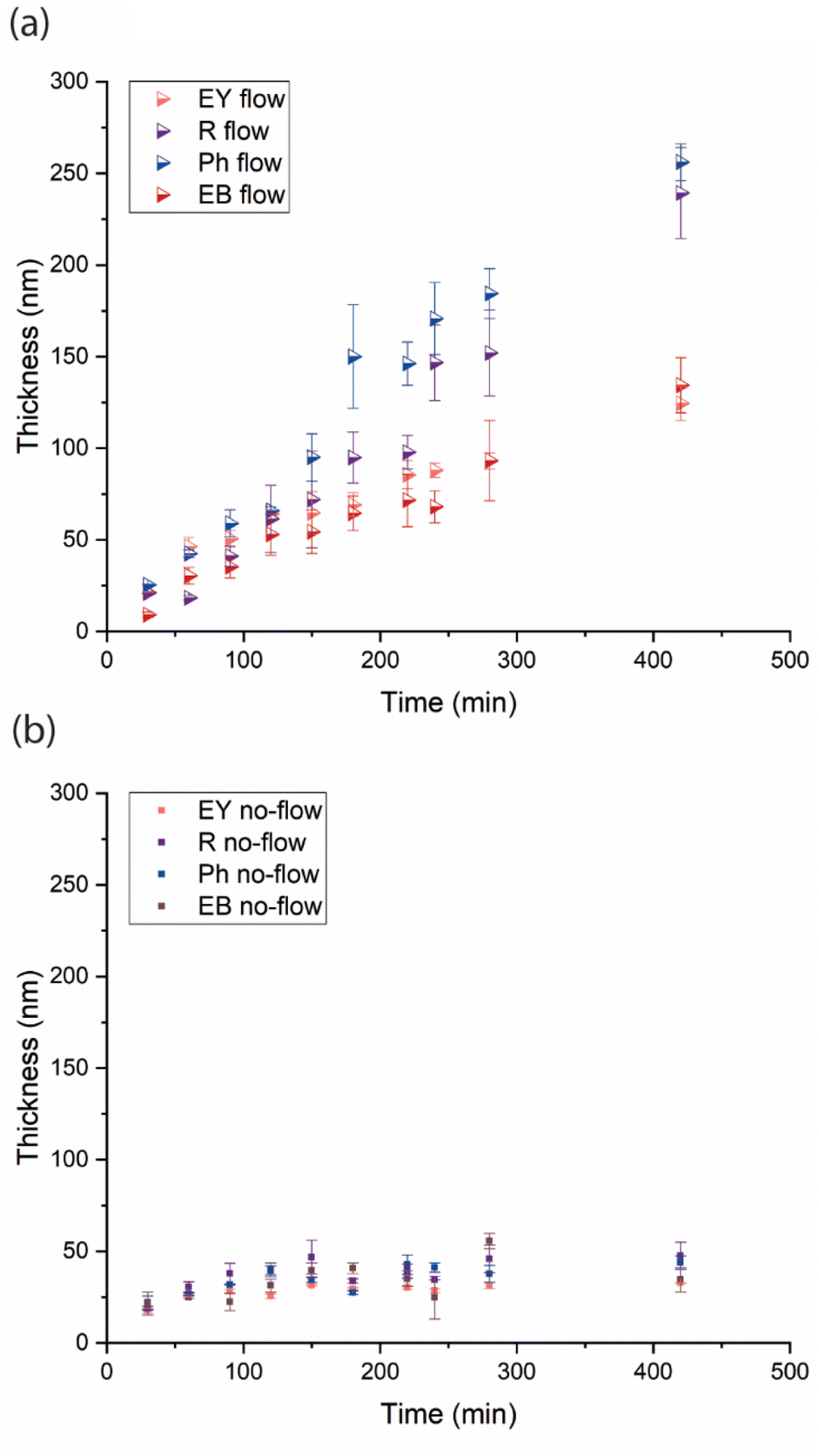 | ||
| Fig. 3 Dry thickness of poly(MeOEGMA) brushes as a function of the polymerization time, as determined by ellipsometry in flow (a) and no-flow (b) conditions using different photocatalysts (EY, R, Ph and EB) for the SI-PET-RAFT process. | ||
We observed similar kinetics under flow conditions in the presence of a free-RAFT agent in solution (Fig. S4†). The chemical composition of polymer brushes in no-flow and flow conditions, as far as observable by XPS, are the same. We do observe a marked difference in the efficiency of polymer brush growth in dependence on the photocatalyst structure. The speed of polymerizations in the presence of RB and phloxine Ph is significantly higher than those of EB and EY, which is probably related to the higher stability of those dyes in an oxygen-containing environment, as also previously observed in PET-RAFT flow systems.38
The grafting density and molecular weight of polymer brushes, particularly when obtained by the “grafting from” approach, are often difficult to determine. Typical techniques of molecular weight investigation, such as size exclusion chromatography (SEC) or gel permeation chromatography (GPC), nuclear magnetic resonance spectroscopy (NMR), and static or dynamic light scattering, are not readily accessible for investigation of these surface-bound polymers, as often degrafting does not provide enough material. In addition, the degrafting polymer brushes often damage the molecular structure or oxidize the end groups, ultimately affecting the outcomes. In our study, we thus sought for an alternative, and have used single-molecule force spectroscopy (SMFS) for the determination of the contour length of individual polymer chains, and, consequently, of the molecular weight Mn, grafting density σ, and reduced grafting density Σ. This technique was previously reported by the groups of Barner-Kowollik, Lee-Thedieck42 and Pop-Georgievski,43 and showed a good correlation with results obtained by GPC.
The RAFT agent at the end of the polymer brush was converted to a thiol group using aminolysis.43 These thiol-ended functional groups will readily attach to gold-coated cantilevers during SMFS measurements (all performed in Milli-Q water; Fig. 4a). Between 6000–20000 force–distance curves were measured for each sample, acquired at many points distributed across the sample surface (Fig. 4b and c, and ESI Fig. S5–S20†). From these, we selected curves showing a clear single unfolding and rupture event. The force curves were further fitted using a worm-like chain (WLC)42,44 model (eqn (S1)), to obtain the contour (Lc) and persistence length of the polymer brush. The molecular weight of polymer brushes was calculated from the average contour length.
 | ||
| Fig. 4 (a) Schematic depiction of SMFS measurements. (b and c) Representative force–distance curve (b) and distribution of contour lengths (c) for thiol-terminated poly(MeOEGMA) brushes synthesized by SI-PET-RAFT in no flow conditions, polymerization time 5 min, thickness 7.3 nm. (d) Relationships of dry thickness and Mn of poly(MeOEGMA) brushes obtained by SI-PET-RAFT, using EY as photocatalyst in flow and no-flow conditions. | ||
The results of the SMFS study are summarized in Fig. 4 (see ESI in detail overview Tables S36, S37, and Fig. S5–S20†) and indicated a clear correlation between the molecular weight Mn and the dry thickness of the polymer brush (Fig. 4d). This confirms that the dry thickness of the polymer on the surface and its linear evolution of over time during the polymerization process in SI-PET-RAFT systems are good indicators the overall control of the polymerization.
We also note that chain ends of polymers with a high molecular weight and a high contour length are, on average, closer to the surface. As a result, they have a higher chance to be picked up by the golden cantilever. Consequently, this method will oversample the longer polymers with higher molecular weight. To partially counteract this effect, we mainly focused the SMFS investigations on the initial stage of the polymerization after 5 min (most points), with additional data at 60 min and 120 min. Typically at the beginning of the polymerization, the difference between the polymer chain length and accessibility of the end groups will not be that high.
The average grafting density polymer chains (σ) in no-flow and flow conditions were determined to be at 0.07 ± 0.01 and 0.08 ± 0.01 polymer chains per nm−2, respectively (by equation σ = h·ρ·NA/Mn, where ellipsometry (h), the bulk density of poly(MeOEGMA) was taken to be 1.05 g cm−3, and NA is Avogadro's constant). As can be seen, the grafting density for brushes obtained under flow and no-flow conditions are similar. This was to be expected, since the grafting density is dominated by the – in both cases identical – initial amount of the RAFT agent on the surface. This is, to a large degree, not influenced by the flow, as it basically relates to the chance of starting the polymer brush formation. The relatively low grafting density is effected by the bulky nature of the monomer.
To determine the packing regime of the surface-bound polymers after the SI-PET-RAFT polymerization, we calculated the reduced grafting density Σ (using Σ = σ·π·Rg′, where Rgis the radius of gyration).8 This value includes the volume of the polymer. The Σ in no-flow and flow conditions was determined to be 31 ± 10 and 77 ± 22, respectively. Since under both conditions Σ ≫ 10, this indicates a polymer brush regime (no-flow conditions) and even a high stretching regime (flow conditions).8
The living nature of the SI-PET-RAFT polymerization under flow conditions should allow the growth of a second polymer block from the poly(MeOEGMA) brush macroinitiator. The poly(MeOEGMA) first block polymerized for 120 min reaching a thickness of 58.0 ± 2.2 nm (by ellipsometry). The resulting surface was then washed with water and ethanol, and dried under a flow of Ar. Next, it was placed back in the flow reactor, after which we pumped – again under irradiation using 410 nm light – a polymerization solution containing N-(2-hydroxypropyl) methacrylamide (HPMA) as monomer, with the molar ratio HPMA:TEOA:EY = 400:1:0.1. We further installed a square mask (5 × 5 mm) on the flow cell. The poly(HPMA) second block attachment was conducted for 120 min, reaching a thickness of 126.1 ± 1.6 nm (by ellipsometry). The thickness of the second block was determined to be 66.8 ± 5.9 nm by AFM – using the pattern that resulted from the use of the mask, and to be 68.1 ± 2.2 nm as measured by ellipsometry (Fig. 5). The AFM phase topography further confirmed the difference in physiochemical properties of the polymer on the surface (Fig. 5c). Qualitatively similar results, although obtained with much lower signal/noise ratios, were obtained using N1 XPS mapping spectroscopy (see ESI Fig. S21†). The high thickness of the second block further confirms the improved livingness of the SI-PET-RAFT in flow conditions. Under no-flow SI-PET-RAFT conditions, we could not achieve thicknesses of the second block higher than 10–15 nm.18 while here the 67–68 nm was not the maximum thickness, but simply the thickness obtained after 120 min. The current data indicate to the improved livingness of the chains, and consequently add to the power of controlled polymerization in flow.
 | ||
| Fig. 5 (a) Optical microscope image of line-patterned thick poly(HPMA) layer poly(MeOEGMA) layer. (b) and (c) AFM height and phase topography of sections. | ||
Conclusions
In summary, we report the first use of SI-PET-RAFT in flow conditions, and demonstrate an evident improvement in polymer brush thickness and polymerization control. The kinetic polymer growth showed linear behavior. The improved livingness of this polymerization was confirmed by creating diblock patterned architectures on the surface, in which the second block can be built with thicknesses much thicker than obtained without flow. Investigation of the molecular weight of the obtained brushes in continuous flow and under no-flow conditions by single-molecule force spectroscopy yielded a linear correlation between the molecular weight of the polymer and the dry thickness of the polymer brushes. We envision that this SI-PET-RAFT in continuous flow conditions is the next step in creating polymer brush coatings with better control of the properties of such brushes. It also opens opportunities for combination with online monitoring (e.g. using real-time QCM-D measurements with a transparent cell) and machine learning to optimize flow conditions.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Wageningen University for continued financial support. MvG is financially supported by the VLAG Graduate School and by the European Research Council (ERC) project CoG-CATCH.References
- K. Parkatzidis, H. S. Wang, N. P. Truong and A. Anastasaki, Chem, 2020, 6, 1575–1588 CAS.
- R. Yin, Z. Wang, M. R. Bockstaller and K. Matyjaszewski, Polym. Chem., 2021, 12, 6071–6082 RSC.
- J. De Neve, J. J. Haven, L. Maes and T. Junkers, Polym. Chem., 2018, 9, 4692–4705 RSC.
- M.-N. Antonopoulou, R. Whitfield, N. P. Truong, D. Wyers, S. Harrisson, T. Junkers and A. Anastasaki, Nat. Chem., 2022, 14, 304–312 CrossRef CAS PubMed.
- C. M. Bates, M. J. Maher, D. W. Janes, C. J. Ellison and C. G. Willson, Macromolecules, 2014, 47, 2–12 CrossRef CAS.
- L. Michalek, L. Barner and C. Barner-Kowollik, Adv. Mater., 2018, 30, 1706321 CrossRef PubMed.
- J. O. Zoppe, N. C. Ataman, P. Mocny, J. Wang, J. Moraes and H.-A. Klok, Chem. Rev., 2017, 117, 1105–1318 CrossRef CAS PubMed.
- W. J. Brittain and S. Minko, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3505–3512 CrossRef CAS.
- G. C. Ritsema van Eck, L. Chiappisi and S. de Beer, ACS Appl. Polym. Mater., 2022, 4, 3062–3087 CrossRef CAS PubMed.
- E. M. Benetti, M. Divandari, S. N. Ramakrishna, G. Morgese, W. Yan and L. Trachsel, Chem. – Eur. J., 2017, 23, 12433–12442 CrossRef CAS PubMed.
- G. Liu, H. Cheng, L. Yan and G. Zhang, J. Phys. Chem. B, 2005, 109, 22603–22607 CrossRef CAS PubMed.
- S. T. Ahmed and D. E. Leckband, Adv. Funct. Mater., 2020, 30, 2000757 CrossRef CAS.
- L. W. Teunissen, A. R. Kuzmyn, F. S. Ruggeri, M. M. J. Smulders and H. Zuilhof, Adv. Mater. Interfaces, 2022, 2101717 CrossRef CAS.
- E. Guazzelli, L. Santarlasci, M. Oliva, C. Pretti, M. Romio, A. Glisenti, E. M. Benetti and E. Martinelli, Eur. Polym. J., 2023, 190, 111998 CrossRef CAS.
- D. Gahtory, R. Sen, A. R. Kuzmyn, J. Escorihuela and H. Zuilhof, Angew. Chem., 2018, 130, 10275–10279 CrossRef.
- E. Roeven, A. R. Kuzmyn, L. Scheres, J. Baggerman, M. M. J. Smulders and H. Zuilhof, Langmuir, 2020, 36, 10187–10199 CrossRef CAS PubMed.
- A. R. Kuzmyn, A. de los Santos Pereira, O. Pop-Georgievski, M. Bruns, E. Brynda and C. Rodriguez-Emmenegger, Polym. Chem., 2014, 5, 4124–4131 RSC.
- A. R. Kuzmyn, L. W. Teunissen, P. Fritz, B. van Lagen, M. M. J. Smulders and H. Zuilhof, Adv. Mater. Interfaces, 2022, 9, 2101784 CrossRef CAS.
- A. R. Kuzmyn, A. T. Nguyen, H. Zuilhof and J. Baggerman, Adv. Mater. Interfaces, 2019, 6, 1900351 CrossRef.
- A. R. Kuzmyn, A. T. Nguyen, L. W. Teunissen, H. Zuilhof and J. Baggerman, Langmuir, 2020, 36, 4439–4446 CrossRef CAS PubMed.
- Y. Ko, V. K. Truong, S. Y. Woo, M. D. Dickey, L. Hsiao and J. Genzer, Biomacromolecules, 2022, 23, 424–430 CrossRef CAS PubMed.
- A. R. Kuzmyn, L. W. Teunissen, M. V. Kroese, J. Kant, S. Venema and H. Zuilhof, ACS Omega, 2022, 7, 38371–38379 CrossRef CAS PubMed.
- E. Wischerhoff, N. Badi, J.-F. Lutz and A. Laschewsky, Soft Matter, 2010, 6, 705–713 RSC.
- Y. Yu, M. Brió Pérez, C. Cao and S. de Beer, Eur. Polym. J., 2021, 147, 110298 CrossRef CAS.
- O. Pop-Georgievski, C. Rodriguez-Emmenegger, A. d. l. S. Pereira, V. Proks, E. Brynda and F. Rypáček, J. Mater. Chem. B, 2013, 1, 2859–2867 RSC.
- R. M. Bielecki, E. M. Benetti, D. Kumar and N. D. Spencer, Tribol. Lett., 2012, 45, 477–487 CrossRef CAS.
- K. J. van der Weg, G. C. Ritsema van Eck and S. de Beer, Lubricants, 2019, 7, 84 CrossRef.
- M. Flejszar, P. Chmielarz, M. Gießl, K. Wolski, J. Smenda, S. Zapotoczny and H. Cölfen, Polymer, 2022, 242, 124587 CrossRef CAS.
- M. Vorobii, A. de los Santos Pereira, O. Pop-Georgievski, N. Y. Kostina, C. Rodriguez-Emmenegger and V. Percec, Polym. Chem., 2015, 6, 4210–4220 RSC.
- E. Rizzardo and D. H. Solomon, Aust. J. Chem., 2012, 65, 945–969 CrossRef CAS.
- M. Zamfir, C. Rodriguez-Emmenegger, S. Bauer, L. Barner, A. Rosenhahn and C. Barner-Kowollik, J. Mater. Chem. B, 2013, 1, 6027–6034 RSC.
- J. Niu, Z. A. Page, N. D. Dolinski, A. Anastasaki, A. T. Hsueh, H. T. Soh and C. J. Hawker, ACS Macro Lett., 2017, 6, 1109–1113 CrossRef CAS PubMed.
- T. Lueckerath, T. Strauch, K. Koynov, C. Barner-Kowollik, D. Y. W. Ng and T. Weil, Biomacromolecules, 2019, 20, 212–221 CrossRef CAS PubMed.
- J. Niu, D. J. Lunn, A. Pusuluri, J. I. Yoo, M. A. O'Malley, S. Mitragotri, H. T. Soh and C. J. Hawker, Nat. Chem., 2017, 9, 537 CrossRef CAS PubMed.
- M. Li, M. Fromel, D. Ranaweera, S. Rocha, C. Boyer and C. W. Pester, ACS Macro Lett., 2019, 8, 374–380 CrossRef CAS PubMed.
- M. Rubens, J. H. Vrijsen, J. Laun and T. Junkers, Angew. Chem., Int. Ed., 2019, 58, 3183–3187 CrossRef CAS PubMed.
- B. Wenn, M. Conradi, A. D. Carreiras, D. M. Haddleton and T. Junkers, Polym. Chem., 2014, 5, 3053–3060 RSC.
- N. Corrigan, L. Zhernakov, M. H. Hashim, J. Xu and C. Boyer, React. Chem. Eng., 2019, 4, 1216–1228 RSC.
- E. van Andel, S. C. Lange, S. P. Pujari, E. J. Tijhaar, M. M. J. Smulders, H. F. J. Savelkoul and H. Zuilhof, Langmuir, 2019, 35, 1181–1191 CrossRef CAS PubMed.
- A. Alvarez-Martin, S. Trashin, M. Cuykx, A. Covaci, K. De Wael and K. Janssens, Dyes Pigm., 2017, 145, 376–384 CrossRef CAS.
- M. P. de Haan, N. Balakrishnan, A. R. Kuzmyn, G. Li, H. M. Willemen, G. Seide, G. C. H. Derksen, B. Albada and H. Zuilhof, Langmuir, 2021, 37, 1446–1455 CrossRef CAS PubMed.
- T. Tischer, R. Gralla-Koser, V. Trouillet, L. Barner, C. Barner-Kowollik and C. Lee-Thedieck, ACS Macro Lett., 2016, 5, 498–503 CrossRef CAS PubMed.
- Y.-M. Wang, A. Kálosi, Y. Halahovets, I. Romanenko, J. Slabý, J. Homola, J. Svoboda, A. de los Santos Pereira and O. Pop-Georgievski, Polym. Chem., 2022, 13, 3815–3826 RSC.
- C. Bouchiat, M. D. Wang, J. Allemand, T. Strick, S. M. Block and V. Croquette, Biophys. J., 1999, 76, 409–413 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00488k |
| This journal is © The Royal Society of Chemistry 2023 |