Epigenetics – relevance to drug safety science
Catherine C.
Priestley
*,
Mark
Anderton
,
Ann T.
Doherty
,
Paul
Duffy
,
Howard R.
Mellor
,
Helen
Powell
and
Ruth
Roberts
Safety Assessment, AstraZeneca R&D, Alderley Park, Macclesfield, Cheshire, SK10 4TG, UK. E-mail: catherine.priestley@astrazeneca.com; Fax: +44 1625 231281; Tel: +44 1625 232435
First published on 16th March 2012
Abstract
Epigenetics describes the study of heritable changes in gene expression that occur in the absence of a change to the DNA sequence. Specific patterns of epigenetic signatures can be stably transmitted through mitosis and cell division and form the molecular basis for developmental stage- and cell type-specific gene expression. Associations have been observed that endogenous and exogenous stimuli can change the epigenetic control of both somatic and stem cell differentiation and thus influence phenotypic behaviours and/or disease progression. In relation to drug safety, DNA methylation changes have been identified in many stages of tumour development following exposure to non-genotoxic carcinogens. However, it is not clear whether DNA methylation changes cause cancer, or arise as a consequence of the transformed state. Toxic agents could act at different levels, by directly modifying the epigenome or indirectly by altering signalling pathways. These alterations in chromatin structure may or may not be heritable but are probably reversible. That said, there is currently insufficient data to support inclusion of epigenetic profiling into pre-clinical evaluation studies. Several international collaborations aim to generate data to determine whether epigenetic modifications are causal links in disease and/or tumour progression. It will only be when an understanding of chemical mode-of-action is required that evaluation of epigenetic changes might be considered. The current toxicological testing battery is expected to identify any potential adverse effects regardless of the mechanism, epigenetic or otherwise. It is recommended that toxicologists keep a close watch of new developments in this field, in particular identification of early epigenetic markers for non-genotoxic carcinogenicity. Scientific collaborations between academia and industry will help to understand inter-individual variations in response to drug and toxin exposure to be able to distinguish between adverse and non-adverse epigenetic changes.
1. Background
Epigenetics refers to the heritable changes in gene expression associated with modifications of DNA or chromatin proteins without alteration of the primary DNA sequence. Regulation of epigenetic states are mediated through a combination of DNA methylation, histone modification, nucleosome remodelling, and non-coding RNA-mediated pathways such as silencing RNA (siRNA) and microRNA (miRNA). These modifications are involved in short-term, lifelong and, possibly, transgenerational modulation and adaptation of genome programming to control a diverse range of cellular functions (including X-chromosome inactivation, tissue-specific gene expression, cellular differentiation, genomic imprinting, chromatin structure and stability, maternal versus paternal allelic expression, and aging).1Unlike the genetic code, the epigenome differs between cell and tissue type in response to shifting environmental, psychosocial and physiological exposures and changes throughout life. For this reason, there is an incomplete understanding of the normal state and dynamic variation between and within individuals let alone the interpretation of perceived epigenetic changes. A strong association has been observed between epigenetic changes and carcinogenesis, specifically for intestinal and colon cancers (reviewed in Feinberg).2 Too little global, genome-wide DNA methylation (hypomethylation) is believed to initiate chromosome instability and activate oncogenes and a malignant cell can have 20–60% less genomic methylation than its normal counterpart.3 Conversely, too much global DNA methylation (hypermethylation) may initiate the silencing of tumour suppressor genes. Specific epigenetic alterations of stem/progenitor cells within a tissue, can affect genes that regulate the expansion of progenitor cells and increase their capacity for self-renewal and pluripotency. Associations have been observed between xenobiotic-induced epigenetic changes within gene promoter regions and carcinogenesis for a number of non-genotoxic carcinogens such as phenobarbital, arsenic, peroxisome proliferators and tamoxifen.4 Based on this data, the topic of epigenetic changes in relation to chemical exposure was selected as a top emerging issue by both the public HESI membership and by the Emerging Issues Subcommittee.5
1.1 What is the question/concern(s) of epigenetics in relation to toxicology?
Evidence for xenobiotic-induced epigenetic changes in non-genotoxic carcinogenesis has implications for the assessment of drug safety. Toxic agents could act at different levels, either directly on enzymes controlling epigenetic changes or indirectly to activate or block signalling pathways causing long-term alterations in chromatin structure that may or may not be heritable. Cell-specific epigenetic variation could result in differential pharmacodynamics, pharmacokinetics, and toxicity of drugs in different tissues. Even within the laboratory setting, subtle differences in environmental conditions could result in inter-individual epigenetic variations. A major challenge for toxicologists is to understand the mechanisms driving inter-individual variations in response to drug and toxin exposure and distinguish between adverse and non-adverse signals. For example, a change in DNA methylation may be advantageous or deleterious and should not simply be equated with toxicity.2. State of the science: epigenetic mechanisms and molecular techniques
In order to understand the toxicological concerns it is important to briefly outline the mechanisms that maintain epigenetic states (reviewed in ref. 6 and 7). Several international collaborations aim to generate data to determine whether epigenetic modifications are causal links in disease and/or tumour progression (Table 1). Due to the diverse array of epigenetic control there is no ‘one test fits all’ approach. As detailed below, most projects are focusing on DNA methylation changes but with adoption of next generation sequencing (NGS) delivering high resolution it is likely that NGS data sets will allow comparison of the different epigenomes, resulting in better comprehension of the role of genetic and epigenetic variation in cell physiology. This screening tool is itself at an early research stage for these types of end points and remains quite expensive and labour-intensive. For a comprehensive review of the available sequencing technologies for epigenomic profiling the reader is referred to a recent article by Martens et al.8 Research is also being performed to assess embryonic stem cells as models for epigenetic screening of chemicals but available data are currently limited.9,10| Project | Organiser | Epigenetic modification | Focus | Ref |
|---|---|---|---|---|
| IHEC: International Human Epigenome Consortium; CNG: The Centre National de Genotypage; MARCAR: Biomarkers and molecular tumour classification for non-genotoxic carcinogenesis; ICGC: International Cancer Genome Consortium; BGI: Beijing Genomics Institute; OBELIX: OBesogenic Endocrine disrupting chemicals: Linking prenatal eXposure to the development of obesity later in life. | ||||
| IHEC | Funding bodies from EU, US, Australia, Canada and Asia | DNA methylation, histone modifications, non-coding RNAs | Coordinate epigenome mapping and characterisation worldwide | http://www.ihec-epigenomes.org |
| Human Epigenome Project | Sanger Institute/Epigenomics AG/CNG | DNA methylation | Methylation variable positions in the human genome | http://www.epigenome.org/ |
| Epigenomics Roadmap Project | NIH | DNA methylation, histone modifications, non-coding RNAs | Producing a public resource of human epigenomic data | http://www.roadmapepigenomics.org/ |
| Epigenesys | EU Funded | DNA methylation, histone modifications, nucleosome positioning, non-coding RNAs | 1. Characterising molecular dynamics of epigenetic systems | http://www.epigenesys.eu/ |
| 2. Linking genotypes to epigenotypes | ||||
| 3. Impact of the environment, development and metabolic signals on the epigenome | ||||
| MARCAR Project | IMI | DNA methylation and histone modifications | Biomarkers for non-genotoxic carcinogenesis | http://www.imi-marcar.eu |
| EPITRON | EU Funded | DNA methylation and histone modifications | 1. Defining the epigenetic alterations by which cancer cells deviate from their normal counterparts | http://epitron.eu |
| 2. Identifying novel therapeutic targets and treatment paradigms from these analyses in order to; | ||||
| 3. Generate novel ‘epi-drugs’ and validate them in suitable model systems | ||||
| CANCERDIP | EU Funded | DNA methylation | Deciphering epigenetic controls that lead to human cancer | http://www.cancerdip.eu/ |
| ICGC | Various | DNA methylation | Profiling genomic, transcriptomic and epigenomic changes in 50 different tumour types and/or subtypes | http://www.icgc.org/ |
| Epitwin | Co-funded – EU, King's College London & BGI | DNA methylation | Co-twin investigations of 20 million CpG sites initially focusing on diseases such as obesity, diabetes, allergies, heart disease and osteoporosis | http://www.epitwin.eu/ |
| OBELIX | EU Funded | DNA methylation | Effects of endocrine disruptor chemicals on obesity development | http://www.theobelixproject.org |
2.1 DNA methylation
DNA methylation is associated with transcriptional silencing due to covalent attachment of a methyl group at carbon 5 of cytosine by DNA methyltransferases (DMNTs) and cofactor S-adenosyl-methionine, to cause disruption to the binding of transcription factors and related gene expression machinery. The process is mediated by a family of methylcytosine-binding proteins including MeCP2.11 In addition to DMNTs, four classes of enzymes establish and maintain DNA methylation patterns: (1) chromatin remodelling enzymes; (2) histone acetyltransferases (HATs); (3) histone deacetylases (HDACs) and (4) histone methyltransferases (HMTs). Establishment of the DNA methylation pattern occurs during development through a dynamic, yet highly regulated process of methylation and demethylation reactions which are activated after the first few hours after conception.12 Importantly, recent reports show that a regulatory protein complex ultimately regulates gene expression, regardless of methylation status.13Detection techniques to differentiate methylated DNA sites are based mainly on digestion with methylation-sensitive restriction enzymes (MSRF) or deamination of non-methylated cytosine residues to uracil with bisulfite treatment, prior to downstream analysis.14–16 These techniques are labour intensive, relatively low throughput and require good DNA integrity.13 Advancement of the bisulfite technique with NGS allows comparative assessment of whole genome methylation patterns. Recently, a further global methylation technique, methylated DNA immunoprecipitation (MeDIP) has been developed which utilises antibodies to the 5′-methylcytosine epitope to enrich methylated DNA.17,18 These fragments can then be individually cloned and hybridised to a tiled genomic microarray (MeDIP-chip) and/or used in NGS (MeDIP-seq). In this respect, these techniques provide an unbiased, genome-wide and gene-specific view of the status of the methylome without a priori selection of candidate genes.
Reporter-type mouse models have been developed with the supposition that the DNA methylation changes identified in the reporter are representative of the entire genome, commonly the viable yellow agouti (Avy) mouse,19 as well as Axin fused (AxinFu)20 and CDK5 activator binding protein-intra-cisternal A particle (CabpIAP)21 models. These mice harbour a metastable gene due to insertion of the IAP transposable element that is normally silenced. In the Avy mouse, when the transposon is unmethylated an alternative start site present within the transposable element results in constitutive ectopic expression of the agouti gene, resulting in yellow mice rather than wild-type brown mice. This model has been employed to investigate the importance of maternal nutrition in determining the susceptibility of offspring to epigenetic (DNA methylation) modification.22 Maternal dietary supplementation during pregnancy with either methyl donating substances, i.e., folic acid, vitamin B12, choline, and betaine,22 or genistein,23 a phytoestrogen present in soy products, shifted offspring coat colour to a higher prevalence of the wild-type phenotype that was directly attributable to cytosine methylation in the agouti-associated transposon.23–26 The use of these models as potential screening tools for chemical safety has recently been questioned27 since it is believed they might be too sensitive and metastable epialleles have not been identified in humans. It was further highlighted that it will be difficult to directly interpret the significance of these data for the whole animal and human due to species differences in epigenetic control mechanisms as well as metabolism and tissue-specific responses.
2.2 Histone modifications
A wide range of posttranslational modifications are associated with amino acid residues in histone tails, namely acetylation, phosphorylation, methylation, demethylation, ubiquitination, biotinylation, sumoylation and ADP-ribosylation to regulate gene transcription, DNA repair and mitotic condensation of chromatin leading to a more open or restricted state28 (Fig. 1). The physiological drivers and regulators of these processes are poorly understood. Acetylation of specific lysine residues within histones is mediated by histone acetyltransferases (HATs). This process is reversed by deacetylation of histone tails by histone deactelyases (HDACs), thus causing gene silencing. Methylation is mediated by histone methyltransferases (HMTs) and this process can be reversed by histone demethylases. Two classes of lysine histone demethylases (HDMs) have been discovered. Lysine demethylase 1 (LSD1) removes mono-/di-methylation whereas the jumonji C-terminal domain (JmjC) family of histone demethylases remove all methylation states.29 Methylation of arginine residues can be antagonised by deimination, a process that uses the enzyme peptidyl arginine deiminase 4 (PADI4) to convert histone arginine to citrulline.30 As previously shown with DNA methylation, specialised nuclear proteins associated with chromatin remodelling complexes, actually work to alter specific DNA regions and histone-DNA interactions.31 It is clear that further work is required to link histone pattern with biological function. Global identification of modified histones has until recently been limited to hybridising antibodies to specific epitopes to enrich methylated histones (chromatin immunoprecipitation, ChIP) and then hybridising onto specific microarrays (ChIP-chip), but low spatial resolution and genomic coverage has limited the output.32 The adoption of ChIP with NGS has led to comprehensive mapping of relatively short DNA sequences giving single base-pair resolution.33 Recent updates from the International Human Epigenome Consortium (IHEC, http://www.ihec-epigenomes.org/) is to map 6 histone modifications (H3K4me3, H3K9me3, H3K27me3, H3K27ac, H3K4me1, and H3K36me3), with plans to extend this number upon discovery of other relevant molecular chromatin features.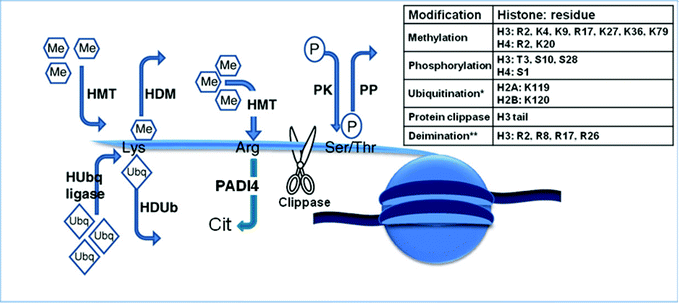 | ||
| Fig. 1 The histone code – the key to the genome. Actions of histone-modifying enzymes. HDM, histone demethylases; HMT, histone methyltransferases; HUbq ligase, histone ubiquitin ligases; HDUB, histone deubiquitinases; PADI4, peptidyl arginine deiminase 4; PK, protein kinase; PP, protein phosphatase; Me, methyl groups; P, phosphate groups. Inserted table shows the residues known to be modified on the various histones. *Ubiquitination refers to the signalling processes mediated through mono-ubiquitination rather than polyubiquitin-mediated proteasomal degradation. **Deimination refers to the process that converts histone arginine to citrulline and antagonizes arginine methylation. Adapted with permission from Best and Carey.34 | ||
2.3 Non-coding RNA-mediated pathways
RNA interference (RNAi) is a process which is highly conserved between organisms whereby double-stranded RNA (transcribed from non-coding DNA) silences transcriptional expression through (a) mRNA degradation and (b) condensation of heterochromatin. The latter mechanism maintains specific chromatin states as well as maintaining genomic integrity by silencing regions of invasive DNA, such as transposon and retroviral elements and heterochromatin regions at centromeres. This effect is associated with formation of short interfering (si)RNA of 21–23 nucleotides in length. The mechanisms of micro-(mi)RNA biogenesis employ the same biochemical reactions that are also responsible for siRNA formation. Each miRNA can be regulated by several miRNAs and one miRNA can recognise several targets.35 Predictions for mRNA-target recognition have been developed using computer modelling. It is believed that expression of 30% of human genes may be regulated by miRNA.36 Impairments in the functioning of miRNA suppressing gene expression at the level of translation have been associated with cancer (reviewed in ref. 37–39). Techniques to analyse miRNA expression have utilised tools from gene expression studies with mRNA, specifically miRNA microarray platforms, qPCR and NGS. Limitations of these techniques are reviewed by Git et al.40 but in short, issues reside with which housekeeper genes to use to normalise the data and the specificity of oligomers due to their short lengths and similarity to miRNA family members.3. Relevance of epigenetic modifications for drug safety science
3.1 The impact of therapeutic modification of the epigenome on toxicity, both target and off-target related changes
Global reduction in DNA methylation and global alterations in histone post-translational modifications have been identified as general features in many steps of tumour development and progression in man. It is thought that DNA methylation contributes to the transformed state by mobilising usually silent transposable elements.41 However, it is not clear whether DNA methylation changes cause cancer, or arise as a consequence of the transformed state.42A number of rodent and human studies have demonstrated that environmental exposure can influence changes in DNA methylation patterns. Specifically, a number of environmental factors, such as famine (Dutch Hunger Winter during World War II), nutritional supplements (folate, vitamin B12, phytooestrogens e.g. genistein), behavioural cues (maternal stress and nurturing), reproductive factors, low-dose radiation, pharmaceuticals (diethystilbestrol, tamoxifen), industrial chemicals (arsenic, bisphenol A) and non-genotoxic carcinogens (Phenobarbital) have all been associated with changes in DNA methylation patterns (reviewed in ref. 43). Common themes were noted from rodent studies using non-genotoxic carcinogens in that global methylation patterns were altered in tumour versus healthy tissues, changes were more pronounced in tumour-prone strains and, where tested, epigenetic changes returned to a normal state following a period of recovery.44 The focus of these studies has presently been on rodent non-genotoxic carcinogens targeting liver but the approach is hoped to facilitate predictions for other organs. However, as kidney and bone marrow are likely to be the next tissues to be studied, their complexity is much greater than the liver in terms of the variety of specialised zones and cells, making the epigenetic states complex. Two major issues remain, firstly determining if the change is adaptive or adverse and secondly ascribing the mechanism as epigenetic.
The key molecular events linking epigenetic change to phenotypic effects could in part be due to (1) inhibition of the enzymes that regulate DNA methylation and histone modifications, reflecting global, genome wide mis-regulation of DNA methylation and/or histone modifications and/or (2) chromatin alteration of individual gene-promoters, specifically growth-promoting and tumour-supressor genes in carcinogenesis associated with aberrant genome regulation. Indeed, genetic mutations of chromatin remodelling genes have been shown in chronic myeloid leukaemia, implying that mis-regulated remodelling can lead to mis-regulated genes.45 In addition, enzyme inhibition of DMNT and HAT by carcinogenic metals, cadmium and nickel, respectively caused a global decrease in epigenetic status.46,47 These enzymes have been successfully targeted by a number of small molecule drugs to inhibit epigenetic modifications, thus re-activating tumour-supressor genes or genes that increase the sensitivity of the target tissues to anticancer drugs. To date, epigenetic drugs tested in clinical trials have focused on reversal of aberrant epigenetic states associated with haematological cancer by targeting DNA methylation (DNMTs) and histone deacetylation (HDACs) enzymes, but have shown little efficacy against solid tumours. Currently, four epigenetic drugs are available for clinical use, two DNA demethylating agents, 5-azacytidine (Vidaza, Celgene) and decitabine (Dacogen, Eisai), both approved for myelodysplastic syndrome, and two histone deacetylase inhibitors, vorinostat (Zolinza, Merck) and romidepsin (Istodax, Celgene) both approved for cutaneous T cell lymphoma. Primary toxicological concerns were for widespread gene expression changes during reproduction and embryonic development, and for carcinogenesis due to their extensive functions in cell growth, differentiation and survival. However, until now clinical trials have mostly shown manageable side effects (nausea, diarrhoea, vomiting and pyrexia). Cardiotoxicity,48 thrombocytopenia49 and teratogenic effects50 have also been observed with some HDAC inhibitors. It is still unclear whether these toxicities are target- or chemistry-based or associated with the treatment population.
Off-target HDAC inhibition has been linked to the teratogenic effects of valporic acid (an anti-epileptic) in mice50 as well as DMNT1 inhibition by hyaldrazine (anti-hypertenisive) and procainamide (antiarrhythmic) triggering a lupus-like autoimmune disease.51 A number of association studies have been or are being conducted on pharmaceuticals, linking off-target epigenetic changes with toxicity via (a) direct inhibition of epigenetic machinery e.g. hydralazine52 or (b) indirect induction through interaction with cellular machinery e.g. thalidomide,53 tamoxifen,54 fluoxetine55 and imipramine.56
3.2 Transgenerational effects of environmental and xenobiotic-induced epigenetic perturbations
There are two major periods of epigenetic reprogramming during mammalian development, occurring early in embryogenesis (preimplatation) to remodel the epigenome following fertilisation and late in embryogenesis to reprogram specifically primordial germ cells when they enter mitotic (male) or meiotic (female) arrest. Genomic imprinting is the best characterised epigenetic event in early mammalian development and results in monoallelic gene expression and X-chromosome inactivation. It is well known that dysregulation of imprinted genes during early development is involved in disorders such as Angelman's, Prader–Willi and Beckwith–Wiederman syndromes, certain cancers, and possibly in autism and other neurological syndromes.57 Of particular interest to developmental and reproductive toxicologists is whether toxic exposure can directly alter the epigenome during early development and whether these alterations are deleterious, presenting a potential hazard.In order for xenobiotic-/environmental-induced epigenetic changes to be classed as transgenerational they require changes to be inherited through the germline from parent to child (F1) and transmitted to subsequent generations, (F2+). Two examples commonly cited include the effects of hormonally active chemicals diethylstilbestrol (DES) (reviewed in Crews and McLachlan58) and vinclozolin.12,59 Exogenous in utero exposure to DES (synthetic oestrogen) induced reproductive disorders and uterine tumours in F2 and F3 generations in humans and mice with alterations in DNA methylation of certain genes under the control of oestrogen receptors (e.g. lactoferrin and c-fos), as well as histone acetylation at developmental genes of the WNT signalling pathway.60,61 Male rats exposed to endocrine disruptors, vinclozolin or methoxychlor during the period of gonadal sex determination had reduced sperm count and viability and increased rates of infertility in adulthood for four generations with associated methylation changes that were stably transmitted through the male germline.62 Both studies comment on the associated epigenetic changes but make no attempt to investigate mechanistically how the epigenetic changes link with the observed reproductive phenotype. It is still unclear from these studies whether epigenetic mechanisms are a cause or consequence of loss of gene expression. What can be said is that the altered epigenotype correlated well with the heritable effects on reproduction but in terms of risk assessment the observed phenotypic trait is qualification of toxicity.
3.3 Contribution of the underlying epigenetic status in models of toxicity to variation in drug response
As mentioned previously, epigenetic changes occur throughout life and differ between species, sex, strains, organs, tissues and cells. It is likely that inter-individual variability in pre-clinical models could result not only from genetic differences but from exposures to different behavioural conditions, diets and chemicals at critical periods throughout life, causing changes in epigenetic status. Even imprinted genes, the most highly conserved in mice versus man, have shown a high level of discordance and dissimilarity. One specific example is the tumour suppressor gene M6P/IGF2R which is imprinted in rodents and expressed from the maternal allele after embryonic implantation, whereas both alleles are functional in humans. Hence, only one rather than two alleles needs to be mutated in rodents to completely inactivate the function of this tumour suppressor gene.63 This is just one example where species difference has implications in carcinogenic risk assessment between rodent and man.Epigenetic factors are increasingly thought to play a role in the inter-individual variability observed in drug metabolism, in particular that involving the cytochrome P450 (CYP) class of drug metabolising enzymes. This variation in drug metabolism can, in turn, influence the clinical outcome of drug treatment, resulting in toxicity or non-response, in particular of anticancer treatments that rely on metabolism for their activation. Although genetic polymorphisms (single nucleotide polymorphisms, insertions/deletions and copy number variants) account for the variability seen in CYP2C9, CYP2C19 and CYP2D6,64 the variability in CYP1A2 and CYP3A4, both of which play key roles in drug metabolism, cannot be explained by genetic polymorphisms. As it is unlikely that major genetic variants in CYPs have not yet been discovered, the remaining heritable variability may be due in part to epigenetic changes in CYPs, as well as in the genes that control their expression.
CYP1A2 is not only one of the most abundant CYPs but an increasing number of drugs are metabolised by this enzyme. Although expression of CYP1A2 can be regulated by environmental factors such as smoking and the consumption of certain foodstuffs, the high inter-individual variability in CYP1A2 activity cannot be accounted for by this alone and no genetic polymorphisms have been identified that can account for this discrepancy. Epigenetic regulation of CYP1A2 is evident from two studies showing that methylation of single CpG islands adjacent to the AP-1 binding site in the 5′-flanking region of the gene65 and in exon 266 have been associated with the regulation of CYP1A2 expression. Methylation status is also thought to be important in the regulation of CYP3A4 expression but in this case it is not methylation of the CYP3A4 gene itself but that of PXR, a key transcription factor involved in the regulation of CYP3A4.67 DNA methylation of the CpG-rich sequence of the PXR promoter was inversely related to CYP3A4 expression and treatment with 5-aza-dC, which induced hypomethylation of the PXR promoter, is associated with increased expression of CYP3A4 mRNA. Methylation status has also been demonstrated to be important in the regulation of a number of other CYPs which play lesser roles in drug metabolism but are thought to be important in carcinogenesis such as CYP1A1, CYP2A13, CYP1B1 and CYP2E1 (reviewed by Rodriguez-Antona et al.68). In addition, chromatin and histone modification has also been suggested to play a role in the regulation of CYP1A1.69
4. Toxicity testing strategies
Toxicity testing of pharmaceuticals aims to identify hazard(s) and subsequently characterise risk. The phenotypic outcomes are the endpoint irrespective of the mode of action, epigenetic or otherwise. The current genotoxicity testing strategy includes a mammalian cell gene-mutation assay (usually at the thymidine kinase locus) that is able to detect mutation events induced by a genetic mutation or epigenetic silencing of the allele. Likewise, traditional and transgenic cancer bioassays examine grossly and histopathogically for tumours irrespective of the mode of action of the test substance. Agents capable of inducing tumours through epigenetic modifications will be identified in these studies. That said, understanding mechanisms of action can help steer drug discovery projects and identify biomarkers of effect to aid subsequent candidate programmes. However, the question remains ‘When is the right time to include epigenetic screening into toxicity testing?’ (Fig. 2). Further, alteration of DNA methylation in a rodent may not be relevant to humans because human cells have a greater capacity for maintaining normal methylation status than those of the rodent.70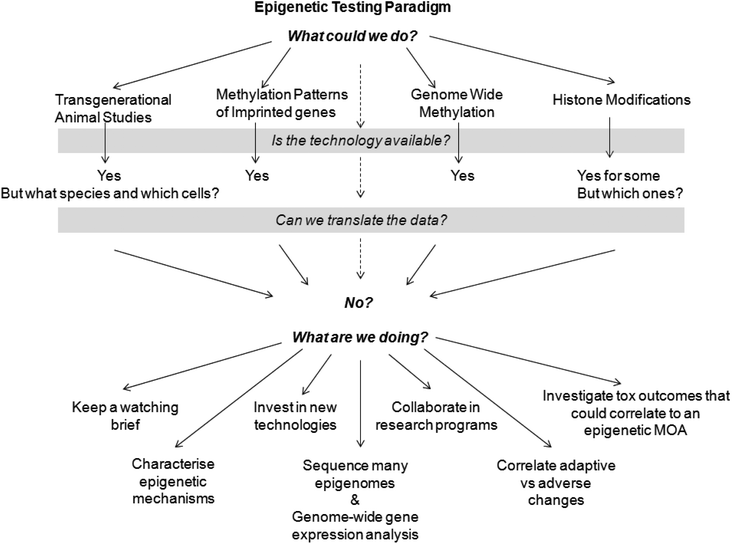 | ||
| Fig. 2 Epigenetic testing paradigm. | ||
To assess the relevance of epigenetic modifications identified in vitro for drug safety science, it is essential to investigate such changes in putative target organs in vivo. It is worth bearing in mind however that some epigenetic modifications are likely to occur normally, and that caution should be taken before ascribing such changes as a toxic response a priori.71 It is therefore essential to establish both dose response, particularly at low exposures, and reversibility, to aid risk assessment and dose setting. Watson and Goodman71 suggested that incorporating the assessment of epigenetic modification status into the overall safety package may enhance and streamline the process by facilitating the ability to (1) detect potential toxicity early (e.g. during in vitro studies and/or subchronic evaluations in vivo), (2) select rational doses for testing, especially in vivo, rather than placing excessive reliance upon doses that are too high, (3) define the shape of the dose-response curve and establish if toxicity only occurs at doses that result in epigenetic modification in the target organ, and/or (4) select appropriate doses for extrapolation across species. The fact that some epigenetic modifications such as histone acetylation, methylation and ubiquitination have been demonstrated to be reversible (as reviewed by Reamon-Buettner and Borlak6), not only offers hope for epigenetic therapies but also raises the possibility that drug-induced epigenetic changes are reversible.
The role of epigenetics in drug safety science was the topic of two recently held workshops by ILSI-HESI27 and National Academy of Sciences.72,73 Several common themes were highlighted; (1) incomplete understanding of the epigenetic processes with key data and knowledge gaps; (2) uncertainty of long-term effects to public health; (3) insufficient validation of epigenetic tests for inclusion into the regulatory process, specifically, no single test is ideal for all epigenetic effects. Clearly for DNA methylation, histone modifications, and miRNA there are many differences between cells, tissues, organs, individuals, strains and species that are essential to normal biological processes in differentiation and development, but there is little understanding of the consequences of inhibition of epigenetic systems (both adaptive and adverse). Without a complete understanding of normal variability it is difficult to make conclusions from any observed changes in the epigenome. Thus, to establish a screening cascade, a number of questions would need to be answered including the following; What endpoint or mechanism would such a screen target in the epigenetic processes? How would false and true positives/negatives be distinguished (adaptive versus adverse)? What follow-up testing strategies would be employed that would be relevant for predicting risk to man? Current testing strategies have focused on DNA methylation changes as these have been shown to be among the earliest observed molecular events in tumour development. The premise that identification of signature patterns of methylated DNA hotspots may be used as early biomarkers for toxicity. Data demonstrating that a chemical is acting by a secondary mechanism can be used to set a safe exposure level. The some-risk-at-all-dose assumption typically made for genotoxic carcinogens is not required when the induction of tumours is a consequence of a classical toxic event seen only at a threshold of exposure.74 Combinational profiling with DNA methylation and gene expression may give an integrative view. This approach has been adopted by Novartis with the aim of profiling tissues from pre-clinical animal models to identify early mechanism-based biomarkers for non-genotoxic carcinogenesis. Data from a recent study showed limited correlation between promoter-based DNA methylation and transcriptional changes following 4-week exposure of mice to phenoarbital, a non-genotoxic liver carcinogen.75 Changes in methylation status may precede gene expression changes but as altered methylation is potentially reversible, a change in DNA methylation should not be simply interpreted as an indication of potential toxicity. Additionally, treatment with a single compound has the potential to result in differing epigenetic signatures in different cell types. Taken together with the fact that each pharmacologically active drug will have a set of pharmacology-related genes irrespective of the toxicological profile, this considerably decreases the predictive power of the integrative approach.
Identifying which model systems might be employed to evaluate epigenetic changes affecting the F1 and/or F3 generation was a topic of debate at the ILSI-HESI meeting on epigenetics.27 The group discussed the use of the agouti mouse but concluded these models may be over sensitive to test agents due to the presence of a metastable epiallele which has not been identified in humans. It was thought that as the epigenetic background is pliable and reversible (changes with age, strain, etc.) a well characterised strain should be used with age matched controls. The mouse would be the preferred choice over the rat or rabbit as it is currently the best characterised for evaluating epigenetic change. Alternative models were discussed including zebrafish, Caenorhabitis elegans, honeybees, Drosophila and embryonic stem cells but as the epigenetic control mechanisms across species are not known, in addition to there being metabolism and tissue-specific differences it would be difficult to interpret directly the significance of these data for humans. Additionally, the in vitro models would not evaluate transgenerational effects.
5. Conclusions and recommendations
This review probably raises more questions than it answers, which is a reflection of the state of the science and its application in the risk assessment of pharmaceuticals. Unfortunately, until key data are generated it will be difficult to associate observed toxicity as a direct consequence of epigenetic changes. A clear understanding of the fundamental control of gene expression and the mechanisms that generate and maintain epigenetic states will be required. Indeed this is being addressed by a number of researchers and institutions and a primary focus of the NIH Roadmap Initiative on Epigenomics. Further, there needs to be a better understanding and characterisation of the relationship between epigenetic changes (either adverse or adaptive) and adverse health outcome. In this context, it will be essential to understand the normal variability associated with epigenetic modifications and the influence of experimental variability that might confound data interpretation.The current toxicological testing battery is expected to identify any potential adverse effects regardless of the mechanism, epigenetic or otherwise. As more data are needed to distinguish epigenetic changes as cause or effect of the toxicological outcome, it is not recommended that epigenetic measures be incorporated into risk assessment strategies at present. It will only be when an understanding of chemical mode-of-action is required that an evaluation of epigenetic change is likely. Due to the diverse array of epigenetic control, along with the potential simultaneous occurrence of both genetic mutations and epigenetic modifications, there is no ‘one test fits all’ approach. It will be important for Safety Assessment to contribute to the advancement of validated risk-based approaches, prior to deploying these emerging tools in an epigenetic screening program. As gene regulation is notoriously complex, multiparametric, non-linear and time-dependent it is perhaps naive to consider introducing an epigenetic screening program in isolation. Development capabilities within industry should be facilitated through collaborations to address individual susceptibility, persistence and modality of epigenetic mechanisms in relation to toxicity.
References
- B. E. Bernstein, A. Meissner and E. S. Lander, The mammalian epigenome, Cell, 2007, 128, 669–681 CrossRef CAS.
- A. P. Feinberg, The epigenetics of cancer etiology, Semin. Cancer Biol., 2004, 14(6), 427–432 CrossRef CAS.
- M. Esteller, M. E. Fraga, M. Guo, J. Garcia-Fonciilas, I. Hedenfalk, A. K. Godwin, J. Trojan, C. Vaurs-Barriere, Y. J. Bignon, S. Ramus, J. Benitez, T. Caldes, Y. Akiyama, Y. Yuasa, V. Launonen, M. J. Canal, R. Rodriguez, G. Capella, M. A. Peinado, A. Borg, L. A. Aaltonen, B. A. Ponder, S. B. Baylin and J. G. Herman, DNA methylation patterns in hereditary human cancer mimics sporadic tumorigenesis, Hum. Mol. Genet., 2001, 10, 3001–3007 CrossRef CAS.
- I. P. Pogribny, I. Rusyn and F. A. Beland, Epigenetic aspects of genotoxic and non-genotoxic hepatocarcinogenesis: studies in rodents, Environ. Mol. Mutagen., 2008, 49(1), 9–15 CrossRef CAS.
- HESI, 2010, http://www.hesiglobal.org/files/public/2010%20Annual%20Meeting/Presentations/EI/05-Augustine-Epigenetics.pdf.
- S. M. Reamon-Buettner and J. Borlak, A new paradigm in toxicology and teratology: altering gene activity in the absence of DNA sequence variation, Reprod. Toxicol., 2007, 24(1), 20–30 CrossRef CAS.
- M. Szyf, The dynamic epigenome and its implications in toxicology, Toxicol. Sci., 2007, 100(1), 7–23 CrossRef CAS.
- J. H. Martens, H. G. Stunnenberg and C. Logie, The decade of the epigenomes?, Genes Cancer, 2011, 2(6), 680–687 CrossRef CAS.
- A. M. Osman, D. A. van Dartel, E. Zwart, M. Blokland, J. L. Pennings and A. H. Piersma, Proteome profiling of mouse embryonic stem cells to define markers for cell differentiation and embryotoxicity, Reprod. Toxicol., 2010, 30(2), 322–332 CrossRef CAS.
- J. E. Trosko and C. C. Chang, Factors to consider in the use of stem cells for pharmaceutic drug development and for chemical safety assessment, Toxicology, 2010, 270(1), 18–34 CrossRef CAS.
- A. Dhasarathy and P. A. Wade, The MBD protein family-reading an epigenetic mark?, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2008, 647(1–2), 39–43 CrossRef CAS.
- M. J. LeBaron, R. J. Rasoulpour, J. Klapacz, R. G. Ellis-Hutchings, H. M. Hollnagel and B. B. Gollapudi, Epigenetics and chemical safety assessment, Mutat. Res., Rev. Mutat. Res., 2010, 705(2), 83–95 CrossRef CAS.
- F. Eckhardt, J. Lewin, R. Cortese, V. K. Rakyan, J. Attwood, M. Burger, J. Burton, T. V. Cox, R. Davies, T. A. Down, C. Haefliger, R. Horton, K. Howe, D. K. Jackson, J. Kunde, C. Koenig, J. Liddle, D. Niblett, T. Otto, R. Pettett, S. Seemann, C. Thompson, T. West, J. Rogers, A. Olek, K. Berlin and K. Beck, DNA methylation profiling of human chromosomes 6, 20 and 22, Nat. Genet., 2006, 38(12), 1378–1385 CrossRef CAS.
- P. W. Laird, The power and the promise of DNA methylation markers, Nat. Rev. Cancer, 2003, 3(4), 253–266 CrossRef CAS.
- C. Dahl and P. Guldberg, DNA methylation analysis techniques, Biogerontology, 2003, 4(4), 233–250 CrossRef CAS.
- S. M. Ho and W. Y. Tang, Techniques used in studies of epigenome dysregulation due to aberrant DNA methylation: an emphasis on fetal-based adult diseases, Reprod. Toxicol., 2007, 23(3), 267–282 CrossRef CAS.
- I. Keshet, Y. Schlesinger, S. Farkash, E. Rand, M. Hecht, E. Segal, E. Pikarski, R. A. Young, A. Niveleau, H. Cedar and I. Simon, Evidence for an instructive mechanism of de novo methylation in cancer cells, Nat. Genet., 2006, 38(2), 149–153 CrossRef CAS.
- I. M. Wilson, J. J. Davies, M. Weber, C. J. Brown, C. E. Alvarez, C. MacAulay, D. Schubeler and W. L. Lam, Epigenomics: mapping the methylome, Cell Cycle, 2006, 5, 155–158 CrossRef CAS.
- D. M. Duhl, H. Vrieling, K. A. Miller, G. L. Wolff and G. S. Barsh, Neomorphic agouti mutations in obese yellow mice, Nat. Genet., 1994, 8, 59–65 CrossRef CAS.
- T. J. Vasicek, L. Zeng, X. J. Guan, T. Zhang, F. Costantini and S. M. Tilghman, Two dominant mutations in the mouse fused gene are the result of transposon insertions, Genetics, 1997, 147, 777–786 CAS.
- R. Druker, T. J. Bruxner, N. J. Lehrbach and E. Whitelaw, Complex patterns of transcription at the insertion site of a retrotransposon in the mouse, Nucleic Acids Res., 2004, 32, 5800–5808 CrossRef CAS.
- R. A. Waterland and R. L. Jirtle, Transposable elements: targets for early nutritional effects on epigenetic gene regulation, Mol. Cell. Biol., 2003, 23, 5293–5300 CrossRef CAS.
- D. C. Dolinoy, J. R. Weidman, R. A. Waterland and R. L. Jirtle, Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome, Environ. Health Perspect., 2006, 114, 567–572 CrossRef CAS.
- G. L. Wolff, R. L. Kodell, S. R. Moore and C. A. Cooney, Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice, FASEB J., 1998, 12, 949–957 CAS.
- R. A. Waterland and R. L. Jirtle, Early nutrition, epigenetic changes at transposons and imprinted genes, and enhanced susceptibility to adult chronic diseases, Nutrition, 2004, 20, 63–68 CrossRef CAS.
- C. A. Cooney, A. A. Dave and G. L. Wolff, Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring, J. Nutr., 2002, 132, 2393S–2400S CAS.
- J. I. Goodman, K. A. Augustine, M. L. Cunningham, D. Dixon, Y. P. Dragan, J. G. Falls, R. J. Rasoulpour, R. C. Sills, R. D. Storer, D. C. Wolf and S. D. Pettit, What do we need to know prior to thinking about incorporating an epigenetic evaluation into safety assessments?, Toxicol. Sci., 2010, 116(2), 375–381 CrossRef CAS.
- A. Vaquero, A. Loyola and D. Reinberg, The constantly changing face of chromatin, Sci. Aging Knowledge Environ., 2003, 14, RE4 Search PubMed.
- H. Hou and H. Yu, Structural insights into histone lysine demethylation, Curr. Opin. Struct. Biol., 2010, 20(6), 739–748 CrossRef CAS.
- G. L. Cuthbert, S. Daujat, A. W. Snowden, H. Erdjument-Bromage, T. Hagiwara, M. Yamada, R. Schneider, P. D. Gregory, P. Tempst, A. J. Bannister and T. Kouzarides, Histone deimination antagonizes arginine methylation, Cell, 2004, 118(5), 545–553 CrossRef CAS.
- J. K. Kim, M. Samaranayake and S. Pradhan, Epigenetic mechanisms in mammals, Cell. Mol. Life Sci., 2009, 66(4), 596–612 CrossRef CAS.
- B. Wold and R. M. Myers, Sequence census methods for functional genomics, Nat. Methods, 2008, 5, 19–21 CrossRef CAS.
- G. Robertson, M. Hirst, M. Bainbridge, M. Bilenky, Y. Zhao, T. Zeng, G. Euskirchen, B. Bernier, R. Varhol, A. Delaney, N. Thiessen, O. L. Griffith, A. He, M. Marra, M. Snyder and S. Jones, Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing, Nat. Methods, 2007, 4(8), 651–657 CrossRef CAS.
- J. D. Best and N. Carey, Epigenetic opportunities and challenges in cancer, Drug Discovery Today, 2010, 15, 65–70 CrossRef CAS.
- D. P. Bartel, MicroRNAs: genomics, biogenesis, mechanism, and function, Cell, 2004, 116, 281–297 CrossRef CAS.
- B. P. Lewis, C. B. Burge and D. P. Bartel, Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets, Cell, 2005, 120(1), 15–20 CrossRef CAS.
- G. A. Calin and C. M. Croce, MicroRNAs and chromosomal abnormalities in cancer cells, Oncogene, 2006, 25, 6202–6210 CrossRef CAS.
- G. A. Calin and C. M. Croce, MicroRNA signatures in human cancers, Nat. Rev. Cancer, 2006, 6, 857–866 CrossRef CAS.
- A. Esquela-Kerscher and F. J. Slack, Oncomirs – microRNAs with a role in cancer, Nat. Rev. Cancer, 2006, 6, 259–269 CrossRef CAS.
- A. Git, H. Dvinge, M. Salmon-Divon, M. Osborne, C. Kutter, J. Hadfield, P. Bertone and C. Caldas, Systematic comparison of microarray profiling, real-time PCR, and next-generation sequencing technologies for measuring differential microRNA expression, RNA, 2010, 16(5), 991–1006 CrossRef CAS.
- A. N. Carnell and J. I. Goodman, The long (LINEs) and the short (SINEs) of it: altered methylation as a precursor to toxicity, Toxicol. Sci., 2003, 75(2), 229–235 CrossRef CAS.
- S. Baylin and T. H. Bestor, Altered methylation patterns in cancer cell genomes: cause or consequence?, Cancer Cell, 2002, 1(4), 299–305 CrossRef CAS.
- D. C. Dolinoy, J. R. Weidman and R. L. Jirtle, Epigenetic gene regulation: linking early developmental environment to adult disease, Reprod. Toxicol., 2007, 23(3), 297–307 CrossRef CAS.
- V. Bombail, J. G. Moggs and G. Orphanides, Perturbation of epigenetic status by toxicants, Toxicol. Lett., 2004, 149(1–3), 51–58 CrossRef CAS.
- F. Grand, S. Kulkarni, A. Chase, J. M. Goldman, M. Gordon and N. C. Cross, Frequent deletion of hSNF5/INI1, a component of the SWI/SNF complex, in chronic myeloid leukemia, Cancer Res., 1999, 59(16), 3870–3874 CAS.
- M. Takiguchi, W. E. Achanzar, W. Qu, G. Li and M. P. Waalkes, Effects of cadmium on DNA-(cytosine-5) methyltransferase activity and DNA methylation status during cadmium-induced cellular transformation, Exp. Cell Res., 2003, 286, 355–365 CrossRef CAS.
- L. Broday, W. Peng, M. H. Kuo, K. Salnikow, M. Zoroddu and M. Costa, Nickel compounds are novel inhibitors of histone H4 acetylation, Cancer Res., 2000, 60, 238–241 CAS.
- S. Marsoni, G. Damia and G. Camboni, A work in progress: the clinical development of histone deacetylase inhibitors, Epigenetics, 2008, 3(3), 164–171 CrossRef.
- H. Matsuoka, A. Unami, T. Fujimura, T. Noto, Y. Takata, K. Yoshizawa, H. Mori, I. Aramori and S. Mutoh, Mechanisms of HDAC inhibitor-induced thrombocytopenia, Eur. J. Pharmacol., 2007, 571(2–3), 88–96 CrossRef CAS.
- E. Menegola, F. Di Renzo, M. L. Broccia, M. Prudenziati, S. Minucci, V. Massa and E. Giavini, Inhibition of histone deacetylase activity on specific embryonic tissues as a new mechanism for teratogenicity, Birth Defects Res., Part B, 2005, 74(5), 392–398 CrossRef CAS.
- E. Cornacchia, J. Golbus, J. Maybaum, J. Strahler, S. Hanash and B. Richardson, Hydralazine and procainamide inhibit T cell DNA methylation and induce autoreactivity, J. Immunol., 1988, 140, 2197–2200 CAS.
- C. Arce, B. Segura-Pacheco, E. Perez-Cardenas, L. Taja-Chayeb, M. Candelaria and A. Dueñnas-Gonzalez, Hydralazine target: From blood vessels to the epigenome, J. Transl. Med., 2006, 4, 10 CrossRef.
- P. H. T. Huang and W. G. McBride, Interaction of [glutarimide-2-14C] thalidomide with rat embryonic DNA in vivo, Teratog., Carcinog. Mutagen., 1997, 17, 1–5 CrossRef CAS.
- V. P. Tryndyak, O. Kovalchuk, L. Muskhelishvili, B. Montgomery, R. Rodriguez-Juarez, S. Melnyk, S. A. Ross, F. A. Beland and I. P. Pogribny, Epigenetic reprogramming of liver cells in tamoxifen-induced rat hepatocarcinogenesis, Mol. Carcinog., 2007, 46(3), 187–197 CrossRef CAS.
- N. M. Tsankova, O. Berton, W. Renthal, A. Kumar, R. L. Neve and E. J. Nestler, Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action, Nat. Neurosci., 2006, 9, 519–525 CrossRef CAS.
- A. B. Csoka and M. Szyf, Epigenetic side-effects of common pharmaceuticals: A potential new field in medicine and pharmacology, Med. Hypotheses, 2009, 73(5), 770–780 CrossRef CAS.
- K. S. Sandhu, Systems properties of proteins encoded by imprinted genes, Epigenetics, 2010, 5(7), 627–636 CrossRef CAS.
- D. Crews and J. A. McLachlan, Epigenetics, evolution, endocrine disruption, health, and disease, Endocrinology, 2006, 147(6), S4–S10 CrossRef CAS.
- M. D. Anway, C. Leathers and M. K. Skinner, Endocrine disruptor vinclozolin induced epigenetic transgenerational adult-onset disease, Endocrinology, 2006, 147, 5515–5523 CrossRef CAS.
- R. R. Newbold, R. B. Hanson, W. N. Jefferson, B. C. Bullock, J. Haseman and J. A. McLachlan, Increased tumors but uncompromised fertility in the female descendants of mice exposed developmentally to diethylstilbestrol, Carcinogenesis, 1998, 19, 1655–1663 CrossRef CAS.
- R. R. Newbold, R. B. Hanson, W. N. Jefferson, B. C. Bullock, J. Haseman and J. A. McLachlan, Proliferative lesions and reproductive tract tumors in male descendants of mice exposed developmentally to diethylstilbestrol, Carcinogenesis, 2000, 21, 1355–1363 CrossRef CAS.
- H. S. Chang, M. D. Anway, S. S. Rekow and M. K. Skinner, Transgenerational epigenetic imprinting of the male germline by endocrine disruptor exposure during gonadal sex determination, Endocrinology, 2006, 147(12), 5524–5541 CrossRef CAS.
- J. K. Killian, C. M. Nolan, A. A. Wylie, T. Li, T. H. Vu, A. R. Hoffman and R. L. Jirtle, Divergent evolution in M6P/IGF2R imprinting from the Jurassic to the Quaternary, Hum. Mol. Genet., 2001, 10, 1721–1728 CrossRef CAS.
- M. Ingelman-Sundberg, S. C. Sim, A. Gomez and C. Rodriguez-Antona, Influence of cytochrome P450 polymorphisms on drug therapies: pharmacogenetic, pharmacoepigenetic and clinical aspects, Pharmacol. Ther., 2007, 116(3), 496–526 CrossRef CAS.
- G. J. Hammons, Y. Yan-Sanders, B. Jin, E. Blann, F. F. Kadlubar and B. D. Lyn-Cook, Specific site methylation in the 5′-flanking region of CYP1A2 interindividual differences in human livers, Life Sci., 2001, 69(7), 839–845 CrossRef CAS.
- R. Ghotbi, A. Gomez, L. Milani, G. Tybring, A. C. Syvänen, L. Bertilsson, M. Ingelman-Sundberg and E. Aklillu, Allele-specific expression and gene methylation in the control of CYP1A2 mRNA level in human livers, Pharmacogenomics J., 2009, 9(3), 208–217 CrossRef CAS.
- W. Habano, T. Gamo, J. Terashima, T. Sugai, K. Otsuka, G. Wakabayashi and S. Ozawa, Involvement of promoter methylation in the regulation of Pregnane X receptor in colon cancer cells, BMC Cancer, 2011, 11, 81 CrossRef CAS.
- C. Rodriguez-Antona, A. Gomez, M. Karlgren, S. C. Sim and M. Ingelman-Sundberg, Molecular genetics and epigenetics of the cytochrome P450 gene family and its relevance for cancer risk and treatment, Hum. Genet., 2010, 127(1), 1–17 CrossRef CAS.
- M. Schnekenburger, L. Peng and A. Puga, HDAC1 bound to the Cyp1a1 promoter blocks histone acetylation associated with Ah receptor-mediated trans-activation, Biochim. Biophys. Acta, Gene Struct. Expression, 2007, 1769(9–10), 569–578 CrossRef CAS.
- J. G. Moggs, J. I. Goodman, J. E. Trosko and R. A. Roberts, Epigenetics and cancer: implications for drug discovery and safety assessment, Toxicol. Appl. Pharmacol., 2004, 196(3), 422–430 CrossRef CAS.
- R. E. Watson and J. I. Goodman, Epigenetics and DNA methylation come of age in toxicology, Toxicol. Sci., 2002, 67, 11–16 CrossRef CAS.
- E. Buckley, Epigenetic effects of chemicals not ready for regulatory “primetime”, Pestic. Toxic Chem. News, 2009, 37 Search PubMed.
- B. Hileman, Chemicals can turn genes on and off; new tests needed, scientists say, Environ. Health News, 2009, (2009) August 3 Search PubMed.
- R. J. Scheuplein, The use of biological data in addition to the carcinogen bioassay in quantitative risk assessment, Prog. Clin. Biol. Res., 1995, 391, 347–357 CAS.
- H. Lempiäinen, A. Müller, S. Brasa, S. S. Teo, T. C. Roloff, L. Morawiec, N. Zamurovic, A. Vicart, E. Funhoff, P. Couttet, D. Schübeler, O. Grenet, J. Marlowe, J. Moggs and R. Terranova, Phenobarbital mediates an epigenetic switch at the constitutive androstane receptor (CAR) target gene Cyp2b10 in the liver of B6C3F1 mice, PLoS One, 2011, 6(3), e18216 Search PubMed.
| This journal is © The Royal Society of Chemistry 2012 |