Recovery from applied strain in interpenetrating polymer network hydrogels with ionic and covalent cross-links
Shannon E.
Bakarich
a,
Geoffrey C.
Pidcock
a,
Paul
Balding
a,
Leo
Stevens
a,
Paul
Calvert
b and
Marc
in het Panhuis
*ac
aSoft Materials Group, School of Chemistry, University of Wollongong, Wollongong, NSW 2522, Australia. E-mail: panhuis@uow.edu.au; Tel: +61 24221 3155
bDepartment of Bioengineering, University of Massachusetts Dartmouth, North Dartmouth, MA 02747, USA. E-mail: pcalvert@umassd.edu
cARC Centre of Excellence for Electromaterials Science, University of Wollongong, Wollongong, NSW 2522, Australia. E-mail: panhuis@uow.edu.au; Tel: +61 24221 3155
First published on 3rd September 2012
Abstract
We have prepared an interpenetrating polymer network hydrogel based on the simultaneous formation of two polymer networks, gellan gum and poly(acrylamide). The gellan gum network is ionically cross-linked, while poly(acrylamide) network is covalently cross-linked. These gels can recover 53 ± 4% of the hysteresis of the first compressive cycle and 90 ± 9% of subsequent cycles.
Hydrogels are the so-called ‘soft and wet’ materials that are formed when a highly hydrophilic polymer is cross-linked, forming a porous matrix in which large volumes of water can be accommodated.1,2 Over the past decade these materials have been attracting wide-spread attention as tissue mimics as their elastic moduli is similar to that of many soft tissues.3 However, when highly swollen, most of these hydrogel materials lack the toughness and extensibility to withstand the forces to which common tissues are subjected.4 The weak and brittle nature of these gels was successfully addressed by the introduction of double network (DN) hydrogels by J. P. Gong in 2003.5,6 DN hydrogels are extremely tough synthetic gels consisting of two interpenetrating polymer networks, which are prepared sequentially.5–7 First a single-network hydrogel is prepared by polymerisation of a monomer. This gel is then swelled in a solution of a second monomer, and the second monomer is polymerised resulting in a DN hydrogel. The strongest reported DN gels can withstand 17 MPa in compression and 0.7 MPa in tension.5 Despite their impressive toughness there is an inherent problem with DN, i.e. these gels do not recover from damage done during a loading cycle, resulting in reduced stiffness and strength.6–9 This behaviour is a form of the “Mullins effect” seen in filled rubbers, but with a significant difference related to recoverability.7 In filled rubbers, the sample can recover (typically within an hour), whereas in DN hydrogels the strain causes bond breakage which never recovers. Recent work by the Gong group has demonstrated that anisotropic hydrogels consisting of lamellar bilayers in a polymer matrix can exhibit self-recovery due to the presence of reversible sacrificial bonds.10
Gellan gum (GG) is a naturally occurring anionic polysaccharide synthesised by the bacterium Sphingomonas elodea.11 It has been found to have a variety of commercial applications, including as food additives, controlled release systems for medication, and emulsifiers in skin care products.12 Gellan gum is readily available commercially, and is EU and US FDA approved for food use. Hot solutions of gellan gum readily gel (upon cooling to room temperature) in the presence of mono- or di-valent cations to form firm brittle gels.13 During cooling the biopolymer chains undergo a conformation transition from random coils to double helices which then aggregate to form a gel network.14 The cations promote the aggregation of the double helices through ionic cross-linking, which has been termed “cation-mediated aggregation”.14 Recently, several groups have discussed potential uses of gellan gum hydrogels in regenerative medicine, due both to their mechanical similarity to tissue and to their capability to support viable mammalian cells.15–17
In this paper, we demonstrate that a new type of hydrogel material prepared by the simultaneous formation of two polymer networks exhibits high extensibility and toughness combined with recovery from large strains. Our hydrogels were prepared by combining hot solutions of the biopolymer gellan gum and the monomer acrylamide, which were then cross-linked simultaneously through addition of the relevant cross-linking agents, i.e. CaCl2 and N,N′-methylenebisacrylamide. The resulting hydrogels consisted of interpenetrating polymer networks (IPNs), an ionically cross-linked GG network and a covalently cross-linked synthetic polymer network (Fig. 1).
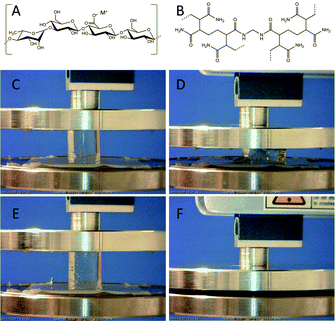 | ||
| Fig. 1 Molecular structure of low-acyl gellan gum (A) and cross-linked poly(acrylamide) (B). (C–F) Typical photographs of the single network gellan gum (C and D) and interpenetrating gellan gum/poly(acrylamide) (E and F) hydrogels prior to compression and after compression to failure. | ||
The concentrations of the reactants (cross-linker, initiator and accelerator) were selected such that polymerisation of poly(acrylamide) (PAAm) in the presence of GG and CaCl2 completed at a room temperature (21 °C) within 2 hours.
The preparation method of the hydrogels (Fig. 1) was as follows. GG hydrogels were prepared from gellan gum solutions (low-acyl gellan gum, CP Kelco, 10 mg mL−1 in Milli-Q water of resistivity 18.2 MΩ cm, 70 °C) cross-linked with CaCl2 (Sigma, 2 mM, 70 °C). The solution was then transferred to a PVC mould to fabricate cylindrical shaped hydrogels. PAAm hydrogels were prepared from a hot (70 °C) solution of acrylamide (AAm, 35 mg mL−1) and N,N′-methylenebisacrylamide (MBAAm, 0.9 mg mL−1) to which N,N,N′,N′-tetramethylethylenediamine (TEMED, 0.094 mg mL−1) and potassium persulfate (KPS, 0.68 mg mL−1) were added prior to transferring to a mould in a desiccator. Following casting, all samples were evacuated to 0.100 bar, to prevent bubble formation, and held for 2 h.
GG/PAAm hydrogels were synthesised by a simultaneous network formation technique. For example, a GG/PAAm hydrogel was prepared by combining hot (70 °C) solutions of MBAAm/AAm and GG, followed by drop-wise addition of hot (70 °C) solutions of TEMED, KPS and CaCl2. The resulting hot solution was transferred to a mould in a desiccator, which was then evacuated (to 0.100 bar) and held for 2 h. The gels with a combination of ionic (I) and covalent (C) cross-links are hereafter referred to as ‘IC’ hydrogels. The water content in these gels was >95% (w/v).
Oscillatory shear rheological measurements were conducted with a controlled strain rheometer (Anton Paar, Physica MCR 301, parallel plate, 21 °C, 45% relative humidity). Strain amplitude sweeps (frequency 10 Hz) revealed that all gels exhibit a clear plateau of storage (G′) and loss (G′′) moduli (Fig. 2). This plateau is commonly referred to as the linear viscoelastic (LVE) region, and the high values of G′/G′′ are indicative of the cross-linked polymer network(s).18 The end of the LVE region represents the shear strain the hydrogels can withstand before the range of inelastic deformation is entered. Comparing the rheological properties of the IC GG/PAAm gels to those of GG gels revealed an increase in G′ (LVE region, from 6.6 ± 1.7 kPa to 8.5 ± 1.1 kPa) and maximum shear strain (from 0.6 ± 0.3% to 2 ± 1%). Frequency sweeps (strain amplitude 0.1%) showed that gellan gum and IC gels exhibited linear dependency of storage modulus on frequency up to 50 Hz and 100 Hz, respectively (data not shown).
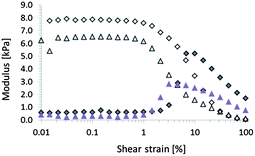 | ||
| Fig. 2 Typical amplitude sweeps (frequency 10 Hz, temperature 21 °C) of single network gellan gum (GG, triangles) and GG/PAAm (IC, diamonds) hydrogels. Open and filled symbols represent storage and loss moduli, respectively. | ||
Compressive stress–strain testing was carried out using a universal testing machine (Shimadzu EZ-S, Japan, cross-head speed 10 mm min−1, 21 °C, 45% relative humidity). Fig. 3a shows a typical stress–strain curve of a GG/PAAm hydrogel with GG and AAm concentrations of 10 mg mL−1 and 35 mg mL−1. At failure, the GG/PAAm hydrogels can withstand 2 and 13 times more stress than the corresponding GG and PAAm hydrogels (see Table 1). The fracture strain of the GG/PAAm hydrogel is 77 ± 3%, which is more than double that of the GG hydrogel and comparable to the PAAm hydrogel. The strain energy to failure of the GG/PAAm hydrogels was 44 ± 6 kJ m−3, which is 29 and 7 times higher than that of the GG and PAAm hydrogels, respectively. These measurements revealed that the GG/PAAm hydrogels exhibit double network behaviour, i.e. the mechanical properties of the GG/PAAm gel were significantly better than those of the gels of the individual components.
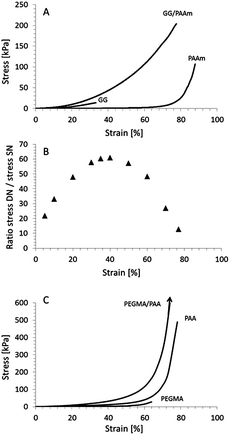 | ||
| Fig. 3 (A) Typical compressive stress–strain curves of single network gellan gum (GG), poly(acrylamide) (PAAm) and IC GG/PAAm hydrogels. (B) Ratio of stress of GG/PAAm hydrogel to GG hydrogel as a function of compressive strain. (C) Typical compressive stress–strain curves (data reproduced from ref. 19) of poly(poly(ethylene glycol) methyl ether methacrylate) (PEGMA), poly(acrylic acid) (PAA) and double network PEGMA/PAA hydrogels. | ||
| Hydrogel | Source | E tan [kPa] | σ max [kPa] | ε max [%] | U [kJ m−3] | U 60 [kJ m−3] |
|---|---|---|---|---|---|---|
| GG | This work | 90 ± 3 | 17 ± 1 | 37 ± 3 | 1.5 ± 0.4 | — |
| PAAm | This work | 1.6 ± 0.2 | 110 ± 6 | 88 ± 2 | 6.4 ± 0.2 | 0.30 ± 0.05 |
| GG/PAAm | This work | 121 ± 10 | 216 ± 31 | 77 ± 3 | 44 ± 6 | 20 ± 3 |
| PEGMA | ref. 19 | 5 | 33 | 63 | 2 | 1.4 |
| PAA | ref. 19 | 31 | 489 | 78 | 34 | 6.3 |
| PEGMA/PAA | ref. 19 | 98 | 7157 | 95 | 663 | 17 |
It has been shown that DN hydrogels show a rapid increase in tangent modulus at high strain values (>75%).5 In contrast, the IC gels exhibit a more gradual increase in modulus leading up to failure. The ratio of the stress in the IC gel to that in the PAAm gel (at equal strain values) exhibits a maximum between 30 and 40% strain, e.g. an improvement of 61 times for the IC, while at failure this ratio reduces to about 2 (Fig. 3b). For comparative purposes we have included the data for a recently published DN poly(poly(ethylene glycol) methyl ether methacrylate)/poly(acrylic acid) (PEGMA/PAA) hydrogel (Fig. 3c and Table 1).19 Analysing the DN data revealed the opposite behaviour, i.e. a small improvement of 18 in the lower strain region, and a significant improvement (266 times) at failure. The stress–strain behaviour of gels with ionic cross-links can be fitted with a power-law, whereas covalently cross-linked gels exhibit exponential behaviour.
It is well-known that DN gels display a large increase in stress at high strain values, typically an increase of an order of magnitude for strain values above 75%.5,19 In contrast, the IC hydrogels can withstand similar stresses as the DN hydrogels for strain values below 60%, despite having failure stresses which are an order of magnitude lower (Fig. 3 and Table 1). This is also evident from the strain energy (0–60%), which is approximately 17 kJ m−3 for the DN and 20 ± 3 kJ m−3 for the IC hydrogel.
Irrespective of their superior strength to other gels the main problem with DN hydrogels is their inability to recover from damage, which has been linked to the breakage of polymer chains in one of the two networks.8 This is where the IC hydrogels have a significant advantage, i.e. one of their networks is cross-linked with ions, which has the ability to reform under ambient conditions.20–22
This was demonstrated using cyclic compression experiments in which the first compressive (virgin) cycle is always performed on a fresh sample that has not been previously deformed. The following cycles are then performed after allowing the sample to rest for a certain period of time. The maximum load in each of the cycles is always 5 N. Analysis was carried out on hydrogels swelled to equilibrium (constant mass) within phosphate buffered saline solution (pH 7.4). Fig. 4a shows typical compression curves with waiting time of 1 h between first and second compressive cycles, 1½ h between second and third cycles and 7 days between third and fourth cycles. The loading–unloading curve of the first compressive cycle shows that the gel has deformed plastically, i.e. the unloading curve follows a different path than the loading curve. This indicates that during compression some of the cross-links have been broken resulting in a weaker gel. However, the loading curve of the second compressive cycle lies between the paths of the loading–unloading curves of the first compressive cycle. This suggests that the cross-links have been partially restored and that the gel is recovering from the applied strain.
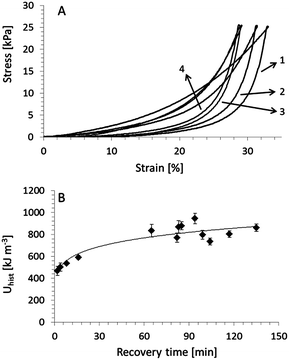 | ||
| Fig. 4 (A) Typical compressive stress–strain curves of gellan gum/poly(acrylamide) hydrogels subjected to a four-cycle experiment with a 1 h, 1½ h and 7 day wait between loading–unloading cycles 1–2, 2–3, and 3–4, respectively. Numbers indicate cycle numbers, cycles strained to a maximum load of 5 N. (B) Hysteresis of loading–unloading cycle 2 (Uhist) as a function of resting (recovery) time between cycles 1 and 2. Uhist values were evaluated using eqn (1). The line is a fit to the data to guide the reader's eye. | ||
The recovery of the gels was analysed using the hysteresis (Uhist, energy dissipated during a cycle), calculated using,8
| Uhist = ∫loadingσdε − ∫unloadingσdε, | (1) |
We observed that although a gel may not be able to fully recover from its first compressive cycle, it can almost fully recover from subsequent cycles. The recoverability is then expressed as a ratio (R2) of a cycle's Uhist to that of the second compressive cycle. For example, the gels recovered to R2 = 78 ± 5% (Uhist = 652 ± 48 kJ m−3) after leaving the gels to rest for 90 min between cycles 2 and 3. This value improves to R2 = 92 ± 6% (Uhist = 750 ± 81 kJ m−3) when left to rest for 1 week. Similar results were obtained for a fourth compressive cycle, i.e. a recoverability of R2 = 90 ± 9%.
In summary, we have prepared hydrogels consisting of an ionically cross-linked biopolymer (gellan gum) and a covalently cross-linked synthetic polymer, poly(acrylamide). These gels displayed double network behaviour, i.e. improved mechanical properties compared to their respective single network hydrogels. The IC hydrogels show that the hysteresis of the first compressive cycle is recovered by 30 ± 2% after waiting for 2 min, and 53 ± 4% after waiting for 80 min between cycles. In contrast, the gels can almost completely recover (with respect to the second compressive cycle) from subsequent loading–unloading cycles. We have suggested that this recovery behaviour can be attributed to the reversible characteristics of the ionically cross-linked biopolymer network in the IC hydrogel. These results contribute to the development of tough hydrogels which can recover from large strains and absorb impacts without permanent damage.
Acknowledgements
This work was supported by funding from the University of Wollongong, and Australian Research Council Centre of Excellence and Future Fellowship (M. in het Panhuis) Programs. Dr R.C. Clark, Mr P. Jackson (both C.P. Kelco), Prof. G. Spinks and Mr D. Kirchmajer (both UoW) are thanked for provision of gellan gum, stimulating discussion, provision of data and technical assistance.Notes and references
- K. Lee and D. Mooney, Chem. Rev., 2001, 101, 1869–1879 CrossRef CAS.
- P. Calvert, Adv. Mater., 2009, 21, 743–756 CrossRef CAS.
- D. Discher, D. Mooney and P. Zandstra, Science, 2009, 324, 1673–1677 CrossRef CAS.
- S. A. Wainwright, W. D. Biggs, J. D. Currey and J. M. Gosline, Mechanical Design in Organisms, Princeton University Press, Princeton, USA, 1986 Search PubMed.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- J. P. Gong, Soft Matter, 2010, 6, 2583–2590 RSC.
- M. A. Haque, T. Kurokawa and J. P. Gong, Polymer, 2012, 53, 1805–1822 CrossRef CAS.
- R. E. Webber, C. Creton, H. R. Brown and J. P. Gong, Macromolecules, 2007, 40, 2919–2927 CrossRef CAS.
- X. Zhao, J. Mech. Phys. Solids, 2012, 60, 319–332 CrossRef CAS.
- M. A. Haque, T. Kurokawa, G. Kamita and J. P. Gong, Macromolecules, 2011, 44, 8916–8924 CrossRef CAS.
- I. Giavasis, L. M. Harvey and B. McNeil, Crit. Rev. Biotechnol., 2000, 20, 177–211 CrossRef CAS.
- A. M. Fialho, L. M. Moreira, A. T. Granja, A. O. Popescu, K. Hoffmann and I. Sá-Correia, Appl. Microbiol. Biotechnol., 2008, 79, 889–900 CrossRef CAS.
- H. Grasdalen and O. Smidsrod, Carbohydr. Polym., 1987, 7, 371–393 CrossRef CAS.
- E. R. Morris, K. Nishinari and M. Rinaudo, Food Hydrocolloids, 2012, 28, 373–411 CrossRef CAS.
- A. M. Smith, R. M. Shelton, Y. Perrie and J. J. Harris, J. Biomater. Appl., 2007, 22, 241–254 CrossRef CAS.
- C. Ferris and M. in het Panhuis, Soft Matter, 2009, 5, 3430–3437 RSC.
- D. F. Coutinho, S. V. Sant, H. Shin, J. T. Oliveira, M. E. Gomes, N. M. Neves, A. Khademhosseini and R. L. Reis, Biomaterials, 2010, 31, 7494–7502 CrossRef CAS.
- T. G. Mezger, The Rheology Handbook, Vincenz, Germany, 2011 Search PubMed.
- S. Naficy, J. M. Razal, P. G. Whitten, G. G. Wallace and G. M. Spinks, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 423–430 CrossRef CAS.
- X. Zhao, N. Huebsch, D. J. Mooney and Z. Suo, J. Appl. Phys., 2010, 107, 063509 CrossRef.
- K. J. Henderson, T. C. Zhou, K. J. Otim and K. R. Shull, Macromolecules, 2010, 43, 6193–6201 CrossRef CAS.
- I. Mora-Barrantes, M. A. Malmierca, J. L. Valentin, A. Rodriguez and L. Ibarra, Soft Matter, 2012, 8, 5201–5213 RSC.
| This journal is © The Royal Society of Chemistry 2012 |