Decoding nonspecific interactions from nature†
Andrew D.
White
,
Ann K.
Nowinski
,
Wenjun
Huang
,
Andrew J.
Keefe
,
Fang
Sun
and
Shaoyi
Jiang
*
University of Washington, Box 351750, Seattle, USA. E-mail: sjiang@u.washington.edu; Fax: +1 206 543 3778; Tel: +1 206 616 6509
First published on 10th September 2012
Abstract
The interactions which govern chemical processes may be broadly categorized into specific interactions, high activity for a certain target molecule, and nonspecific interactions, low activity for all targets. Despite their ubiquity in biology and chemistry, nonspecific interactions are generally overlooked and a fundamental understanding of nonspecific interactions is lacking. Molecular chaperones are large protein complexes which have evolved to resist nonspecific interactions. Their interior surface resists binding to thousands of types of misfolded proteins. Proteins found in the cytoplasm, a crowded environment with many spurious binding targets, are another example. These proteins have evolved high selectivity and stability despite nonspecific interactions. Using structural bioinformatics, we have studied the interiors of molecular chaperones from five species and examined the surface chemistry of 1162 proteins, categorized by if they are present in the cytoplasm or extracellular space. A better understanding of how nature resists nonspecific interactions is key for the chemistry of materials, surfaces, and particles which must remain stable in complex environments. The abundance of amino acids, their interactions, their hydration, and sequence patterns were compared in these two systems, molecular chaperones and proteins surfaces. Striking similarities were found and trends were identified as the system environments became harsher. Peptide based mimics were synthesized to test the conclusions. This, in turn, has led to the design of new stealth compounds and a deeper understanding of nonspecific interactions.
1 Introduction
Specific and nonspecific interactions at interfaces are two extremes governing nearly every biological process and impacting every biological application.1 Specific interactions are high activity interactions between two specific molecules. Extensive research has been performed to study specific interactions such as to design functional peptides through screening libraries of peptides,2 characterize or design proteins possessing activity for a specific target,3 and design catalysts where activity for a specific molecule is desired.4 In contrast, nonspecific interactions are universal interactions for all potential targets and usually the goal is low activity. Examples of systems that emphasize nonspecific interactions include proteins which are stable in environments with many other macromolecules, materials which resist nonspecific binding,5 and enzymes which are highly selective and rarely bind to non-targets. It is fundamentally a challenge to screen or study these ubiquitous and weak nonspecific interactions. Thus, there is a lack of research in this area. One key application of understanding nonspecific interactions is in the development of stealth materials, which resist the attachment of biomolecules and microorganisms. They are invisible to various biological species, hence the term “stealth”. Although many stealth materials, particularly zwitterionic polymers, have been used in applications,5 it is still challenging to design synthetic drug delivery carriers matching native protein circulation time in the blood stream and implantable materials fully compatible with human tissue. For example, the circulation half-life of albumin6 in blood is still longer than any synthetic particles, even after state-of-the-art PEG-modification.7In this work, we turn to nature and use proteins and molecular chaperones as a guide towards understanding nonspecific interactions. Proteins resist nonspecific adsorption in order to be stable in complex environments such as the cytoplasm of a cell, which contains thousands of protein types.8 The cytoplasm is a crowded environment and provides many spurious binding targets for proteins; yet proteins have evolved high selectivity and stability through resisting nonspecific interactions. The non-interacting property of proteins is often put into practice in biomaterials and biosensors research where the protein albumin is used as a blocking agent to block nonspecific adsorption of non-target proteins onto surfaces.9 Another example of nature resisting nonspecific interactions is found in molecular chaperones, which guide proteins from a misfolded or unfolded conformation back into a native conformation.10 The defective (substrate) proteins fold while enclosed inside a cavity of the molecular chaperone. The chemistry of this cavity is unique among biological surfaces in that it contacts not only thousands of proteins, but many conformations of each protein.11 Yet chaperone proteins do not irreversibly bind with proteins. The cavity is sometimes described as a “non-stick” surface.12 Thus molecular chaperones provide a second system which has this non-interacting property.
By examining many proteins from both these systems, it is possible to separate nonspecific effects from the many specific functions of proteins. We use two types of bioinformatics methods for this. The first studies the sequence and abundance of amino acids in the proteins, similar to the molecular formula of a molecule. The second set examines the structure and interactions among the amino acids in a protein and solvent, similar to the 3D structure of a molecule. Through these two methods and two systems, it is possible understand the way these proteins avoid nonspecific interactions. We analyzed a database of protein surfaces and molecular chaperone cavity surfaces using these two techniques. The questions to be answered are which amino acids are most common, how often do they interact, do they interact with water more than other amino acids, and do they prefer to interact with protein surfaces or interiors? Next, the modeling conclusions were used to design peptide based materials which should resist nonspecific interactions. Finally, these peptides were synthesized to test their resistance to nonspecific interactions with proteins. These peptides do create surfaces which resist nonspecific interactions and compare well to other low-fouling peptides which have been reported in the literature.13–16
2 Experimental details
2.1 Protein dataset construction
Three datasets of proteins with Protein Data Bank (PDB) structures were constructed. The first dataset consists of 1162 unique human proteins. The second dataset is a subset of the first with 34 proteins and the additional criterion that the proteins are located in the extracellular space (GO ID: 5615).17 The last is again a subset of the human dataset with the additional criterion of being located in the cytoplasm (GO ID: 5737). It has 221 proteins. We ensure a diversity of structures by using a 40% homologue cut-off and all structures containing residue gaps are excluded. Further constraints are that the X-ray resolution is ≤2.5 Å, ‘mutant’ must not appear in the title, no large ligands (e.g., DNA, RNA), and the only macromolecule in the structure is a protein. The molecular chaperones considered are all crystal structures in the cis or closed conformation. This is the conformation during which substrate proteins refold. The molecular chaperons considered are: GroEL–GroES isolated from Escherichia coli (‘E. coli GroEL’),18 a GroEL–GroES complex isolated from Thermus thermophilus (‘Thermo GroEL–GroES’),19 a group II chaperonin protein isolated from Methanococcus maripaludis (‘Group II’),20 a eukaryotic molecular chaperone isolated from yeast (‘HSP90’)21 and a cytosolic chaperonin isolated from yeast (‘CCT’).22Statistical analysis was performed using the R statistical package.23 SQLShare was used for managing data.24 The PDBs and data used for each dataset are available from: http://sqlshare.escience.washington.edu. The human, cytoplasm, and extracellular datasets are available as SQL data tables under the ‘h2’, ‘cph2’, and ‘eh2’ tags, respectively. X_1, X_2, and X_3 (where X is the dataset) contain the protein information, residue information, and atomic information, respectively. X_c contains the surface contacts (only available for ‘h2’).
2.2 Surface identification
All water was removed from the PDB structures before calculating the accessible surface area. Surface area was calculated using accessible surface area.25 A residue is classified as ‘surface’ when it is 30% or above its maximum surface area. Maximum surface area for a given residue is defined as the surface area occupied by the side chain atoms of a Gly-X-Gly peptide, with X being the residue of interest. The rotamers were taken to be α-helical and the lowest energy χ-rotamers were used.The identification of interior residues in molecular chaperones consists of three steps: (1) identify surface residues, (2) tabulate heavy atoms from the surface residues which are occluded, (3) identify which residues have more than h heavy atoms occluded. Once a residue is identified as a surface residue, it may be either an interior or an exterior surface residue. To be an interior surface residue, h heavy atoms (non-hydrogen atoms), or more, in the residue must not be occluded by atoms from other residues. See the ESI† for the calculation of occlusion and Table S1† for h and other parameters used in the interior identification.
2.3 Structure and interaction equations
The proportion of amino acids which are interacting may be calculated as: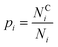 | (1) |
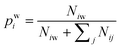 | (2) |
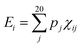 | (3) |
2.4 Peptide synthesis and characterization
N-Fluorenylmethoxycarbonyl (Fmoc)-protected amino acids with the amine and side chain protected (Fmoc–Glu(OtBu)–OH, Fmoc–Lys(Boc)–OH, Fmoc–Cys(Trt)–OH, Fmoc–Pro–OH, Fmoc–Gly–OH), Rink amide AM resin, O-benzotriazole-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU), N-hydroxybenzotriazole (HOBt), and N,N-dimethylformamide (DMF) were bought from AAPPTec (Louisville, KY). N,N-Diisopropylethylamine (DIPEA) was bought from TCI America (Portland, OR). Trifluoroacetic acid (TFA), pyridine, and acetic anhydride were bought from EMD (Darmstadt, Germany). Piperidine, dichloromethane (DCM), triisopropylsilane (TIS), 1,3-dimethoxybenzene (DMB), 1,2-ethanedithiol (EDT), 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP), phosphate buffered saline (PBS), fibrinogen from bovine plasma, and lysozyme from chicken egg white were purchased from Sigma Aldrich (St. Louis, MO). Ethanol was purchased from Decon Labs, Inc. (King of Prussia, PA). Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) was purchased from Pierce (Rockford, IL).The peptides were synthesized using the AAPPTec Titan 357 automated synthesizer by a solid-phase technique, starting from a polystyrene Rink amide AM resin (0.58 mmol g−1 loading capacity). Coupling was performed using amino acid monomer, HBTU, HOBt, and DIPEA prepared in DMF in a molar ratio of 1.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:2 in four times excess of the loading capacity of the resin. Deprotection of Fmoc groups was achieved using 20% piperidine in DMF. N-terminal acetylation was achieved with a solution of pyridine (5%), acetic anhydride (5%) and DMF (90%) (v/v/v). Random peptide sequences were created using the mix and split capability of the AAPPTec Titan 357. The cleavage of the final product was performed with a TFA (75%), DCM (15%), DMB (4%), water (2%), TIS (2%), and EDT (2%) (v/v/v/v) cleavage cocktail. The peptide purity was evaluated by preparative reverse phase high pressure liquid chromatography (RP-HPLC) for known sequences and purified as needed. The purity of the glycine peptide sequences was 92% and the asparagine peptide was 97%. Peptides were analyzed by matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF-MS) (see Fig. S5†).
:1:1:2 in four times excess of the loading capacity of the resin. Deprotection of Fmoc groups was achieved using 20% piperidine in DMF. N-terminal acetylation was achieved with a solution of pyridine (5%), acetic anhydride (5%) and DMF (90%) (v/v/v). Random peptide sequences were created using the mix and split capability of the AAPPTec Titan 357. The cleavage of the final product was performed with a TFA (75%), DCM (15%), DMB (4%), water (2%), TIS (2%), and EDT (2%) (v/v/v/v) cleavage cocktail. The peptide purity was evaluated by preparative reverse phase high pressure liquid chromatography (RP-HPLC) for known sequences and purified as needed. The purity of the glycine peptide sequences was 92% and the asparagine peptide was 97%. Peptides were analyzed by matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF-MS) (see Fig. S5†).
2.5 Peptide self-assembly and protein adsorption
Peptide self-assembled monolayers were prepared as previously described.13 Gold coated surface plasmon resonance (SPR) chips were cleaned by rinsing with Millipore water, ethanol, and then drying with filtered air. They were placed in the UV cleaner for 20 min. Once removed, the gold chips were rinsed again with Millipore water, ethanol, and dried by filtered air. The clean chips were incubated with a phosphate buffered saline (PBS) aqueous solution (pH 7.4 and ionic strength of 150 mM) of 0.2 mg mL−1 peptide for 24 h. After incubation the chips were removed from solution, rinsed with Millipore water, and evaluated by SPR. The SPR sensograms for each condition are shown in Fig. S6.†A laboratory SPR sensor developed at the Institute of Photonics and Electronics, Prague, Czech Republic was used26 as described previously27 to evaluate protein adsorption. Gold chips covered with self-assembling peptides were rinsed with Millipore water, dried by filtered air, and mounted to the device. The temperature controller was set to 25 ± 0.01 °C. Protein adsorption was measured by flowing PBS buffer at 40 μL min−1 for 10 min, 1 mg mL−1 protein solutions of fibrinogen and lysozyme for 10 min, and PBS buffer again for 10 min. The wavelength shift between baselines before protein injection and after buffer rinse was used to quantify the total amount of protein adsorbed. A reference channel containing solely PBS buffer was flown for each chip and its baseline drift was subtracted from the final wavelength change. A 1 nm wavelength shift from 750 nm corresponds to 17 ng cm−2 adsorbed proteins.28 The detection limit for the SPR sensor is 0.2 ng cm−2.27 For statistics reported in this edge article, each chip corresponds to one data point for calculating standard deviations.
3 Results
3.1 Abundance and sequence
After constructing a dataset of proteins found in humans, it is possible to tabulate the most abundant amino acids on protein surfaces. The average fractions of amino acids on protein surfaces found in humans are shown in Fig. 1. The most striking observation is the large fractions of lysine (K) and glutamic acid (E). In fact, charged amino acids, including E, K, arginine (R), aspartic acid (D), and histidine (H), comprise 41% ± 0.3% of the surfaces of proteins. Those amino acids comprise only 27% when considering both surface and non-surface amino acids. It is generally assumed that proteins have the most hydrophilic amino acids on their exteriors and the hydrophobic on the interior in order to maintain their native conformation. Thus the ordering of most common to least common amino acids on the surface may follow how hydrophilic the amino acids are. However, K and E do not have the lowest free energy of solvation;29 hydrophilicity is not the determining property for surface fraction.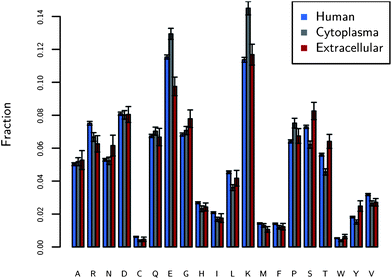 | ||
| Fig. 1 The fraction of amino acids on the surfaces of proteins found in three different locations: human proteins (N = 1162), human cytoplasmic proteins (N = 221), and human extracellular (N = 34) proteins. The y-axis is the median of the fractions of each amino acid over the entire dataset. The figure shows the large fraction of charged residues on protein surfaces, in particular E and K. The error bars are standard errors. | ||
The protein dataset is further broken into the cytoplasm and extracellular. The extracellular environment, the space between cells, generally is not as crowded8 as the cytoplasm and we expect that nonspecific binding is not as interfering compared to the cytoplasm. As seen from our results, the largest difference between cytoplasmic and extracellular proteins is the larger fraction of charged amino acids: 43.8% ± 0.3% for the cytoplasm and 37.5% ± 0.7% for the extracellular. The extracellular dataset is lower in charged amino acids, but higher in polar hydrophilic amino acids (27.3% ± 0.2% vs. 23.7 ± 0.6%), among which specifically serine (S) and threonine (T) are more abundant than those in the cytoplasm. Polar hydrophilic amino acids are S, T, asparagine (N), and glutamine (Q). These results generally follow the trends seen by Andrade et al.,30 who examined a similar though smaller dataset. In the cytoplasm, a crowded environment prone to protein aggregation,8 the proteins have more E and K. Thus, the K and E play an important role in these nonspecific interactions. Evidence that E and K are located in nonspecific regions of protein surfaces (unrelated to function) can be found in work from Jimenez as well, who analyzed protein surfaces broken into regions related to protein function and regions unrelated to function.31 He showed an increase of 36% of charged amino acids in regions unrelated to function relative to regions related to function. See Fig. S1–S3† for a more detailed analysis of the error and sensitivity of these results.
A similar abundance analysis on the interiors of molecular chaperones’ abundance is shown in Fig. 2. The analysis here though is for the interior of a single chaperone protein complex. All the chaperones are in the closed (cis) conformation, during which encapsulated substrate proteins are folding. Again E and K are the most common amino acids in the interior cavities of this collection of chaperone proteins. The fraction of E and K is much larger than for the previous dataset and the fraction of E and K is over 20% on these large proteins. The fraction of charged amino acids, in general, is much higher as well for the molecular chaperones. A dataset containing 528 Escherichia coli (E. coli) protein surfaces was constructed to ensure the results seen on GroEL–GroES are not simply general to E. coli proteins. Also, it is possible to calculate if the fraction of charged amino acids is significant on the chaperone interiors. The fraction of charged amino acids on the surface of E. coli GroEL–GroES is 56%, which is a higher fraction than 98% of all of the E. coli proteins considered (shown in blue in Fig. 2). This large fraction of charged amino acids has been noted before.10,11 However, here we can see exactly which amino acids are more common than expected (E) and how significant it is. Fig. 2 also shows how the interior cavity surface changes between the mesophilic GroEL–GroES and thermophilic GroEL–GroES (optimal growth temperature of 65 °C). Protein folding is typically more difficult at higher temperatures due to the increasing importance of entropy as temperature increases, and thus the thermophilic GroEL–GroES represents a more challenged chaperone. The fraction of charged amino acids is increased to 70% for the thermophilic GroEL–GroES, with most of the increase coming from E and K. Molecular chaperones, where nature requires strong stealth against many protein types, appear to use charged amino acids to accomplish this. E and K are the most utilized charged amino acids in both systems considered.
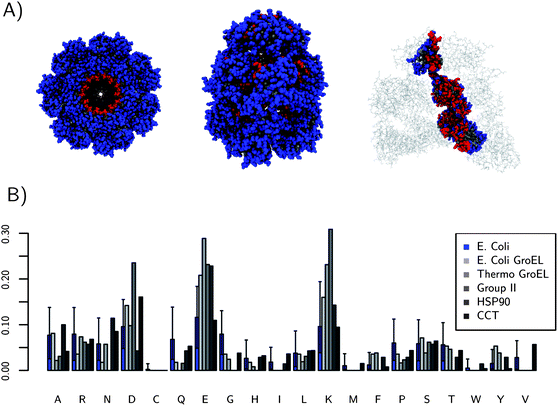 | ||
| Fig. 2 (A) shows three views of the location of interior residues (red) for E. coli GroEL. (B) The fraction of each amino acid on the interiors of 5 molecular chaperones. The median fraction of each amino acid type on the surface of a collection of 528 E. coli proteins is shown in blue for comparison to GroEL–GroES, which is also an E. coli protein. GroEL–GroES is significantly different. All the chaperone structures used for these calculations were the cis or “closed” forms. The error bars come from a 95% confidence interval from quantiling. The fractions of E and K are the most different relative to E. coli proteins. The thermophilic GroEL–GroES mutant has a very high fraction of charged residues, 70%. | ||
The large fraction of K and E suggests that they may be part of a general sequence pattern on protein surfaces; this was found to not be the case. The data considered here are the amino acid pair frequencies in sequence space. The most frequently occurring pairs for the human protein dataset are shown in Fig. 3a. The plot of the pair frequency is in red and the blue shows what the predicted distribution would be if the pairs followed random chance, with a multinomial distribution as a background model. The multinomial model is the number of pairs that would occur by chance if we knew the amino acid surface fractions. Interestingly, few pairs occur next to each other more or less often than the multinomial model suggested within the uncertainty. Results show that on the surface at least, there are no global sequence patterns. There is one exception to this trend in Fig. 3a: the glycine/serine pair. As shown in Fig. S4,† that pair is most commonly found as a type-II turn, resulting in its increased frequency over the multinomial model. This demonstrates that the methodology can discover surface motifs; in this case solvated type-II turns. The large fractions of K and E, however, are not correlated with a frequently occurring sequence motif and are instead nearly randomly distributed.
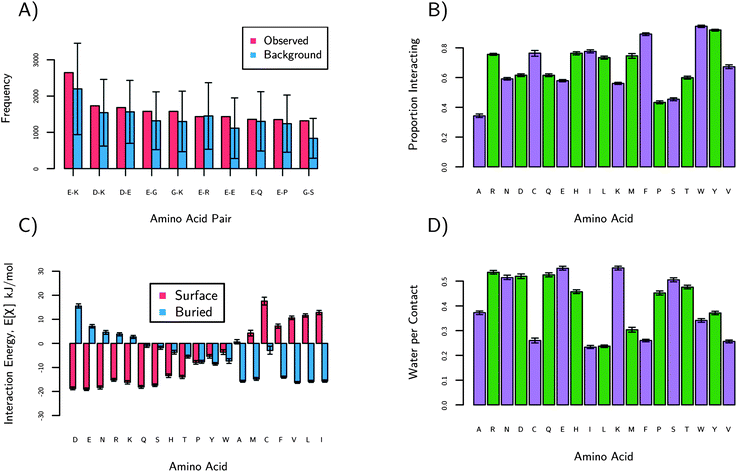 | ||
| Fig. 3 These plots show statistics based on protein sequences (a) and structures (b–d). (a) shows the observed number of pairs of amino acids on the surfaces of proteins. The blue bars are the expected numbers of pairs if the sequence were random. The order of left to right is from most frequently observed to least. Only the G–S pair is considered to be significantly more common than expected. (b) shows the proportion of amino acids interacting from the human dataset. An amino acid is considered as ‘interacting’ if it is in contact with any other amino acid. We see small chains (S, P, A), E and K have the lowest proportions of interactions. (c) shows the preference of each amino acid for protein surfaces or protein interiors. See text for details. D, E, and N have the highest preference for protein surfaces relative to protein interiors. (d) shows the preference of amino acids for water relative to interacting with another amino acids. E and K have the highest water per contact. The green and purple colors are to guide the eyes. | ||
3.2 Structure and interaction
In order to understand why nature chooses E and K, a series of statistical measures based on 3-dimensional structures were calculated and are plotted in Fig. 3b–d. These results are only for the protein surfaces dataset, not the molecular chaperones. Briefly, Fig. 3b shows that E, K and S have very few interactions with other amino acids, Fig. 3c shows that charged (E, K, R, D, H) and amide amino acids (N and Q) have the most disfavored interactions with protein cores, and Fig. 3d shows that charged amino acids, amide (Q, N), and alcohol (S) are the most hydrated. These results are discussed in detail below.Fig. 3b shows how often an amino acid is interacting with any other amino acid, which characterizes their nonspecific interactions. These data are normalized by the number of the amino acids, so that each bar is comparable. These data are only for amino acids observed on the surface. S, K and E have the lowest proportion interacting among the charged and polar hydrophilic amino acids. Alanine and proline are lower due in part to the small size of their side-chains. K and E have large side-chains but still rarely interact. R has the highest proportion of interactions among the hydrophilic amino acids, perhaps explaining why it is so much less often observed on protein surfaces.
In addition to the amount of interactions described above, it is important to discover with what amino acids interact. For nonspecific interactions, reversible interactions are preferred to irreversible. For example, aggregation is often an irreversible process which we expect proteins to disfavor. Therefore we plot the strength of interactions of each amino acid with surface and buried amino acids on an average protein. Generally, more favorable interactions with the buried residues of a protein destabilize the protein, possibly leading to unfolding and aggregation. The average protein is a hypothetical protein which has a surface and buried residue distribution given by Fig. 1. The strength of interactions was calculated using quasi-chemical theory,32 which is described in the ESI.† The results are plotted in Fig. 3c where the red bars indicate strength of interaction between an amino acid and the average protein's surface. The blue bars show the strength of interaction between an amino acid and the average interior of proteins. Amide, charged, and alcohol amino acids have the highest preference for protein surfaces. Combined with our previous results, we see that E and K have fewer interactions (Fig. 3b) and also favor interactions with protein surface amino acids, not interior amino acids (Fig. 3c). Nature seems to prefer these amino acids as well in locations where resisting nonspecific interactions is necessary for function (i.e., the cytoplasm and molecular chaperones).
It is well established that hydration is the key to resisting nonspecific interactions.33,34 Thus, we also analyzed crystallographic proximity of waters to amino acids. The numbers of water per contact are shown in Fig. 3d. The choice of using the number of contacts as the normalization was done to eliminate the size effects of amino acids. Those amino acids with more atoms tend have more waters near them. Therefore, Fig. 3d should be thought of as the preference of an amino acid to interact with water relative to interacting with another amino acid. Again, we see E and K resisting other amino acids and preferring water. The amides and alcohols follow the trend as well. Overall, Fig. 3 shows E and K are randomly distributed and rarely interact. If E and K do interact, they prefer water to amino acids. If E and K do interact with amino acids, they prefer to interact with those found on the surface of proteins instead of the interior. Regardless of the abundances of E and K found, these structural results strongly indicate E and K resist nonspecific interactions.
3.3 Implications for design of stealth materials
There are three separate results indicating E and K resist nonspecific interactions. First, E and K are the most common amino acids on protein surfaces and even more so on protein surfaces in the cytoplasm, a crowded environment. Second, E and K are the most common amino acids found on the interiors of molecular chaperones, a location which must resist nonspecific binding with thousands of protein types and conformations. Lastly, the structure and interaction results indicate that K and E rarely interact with other amino acids, favorably interact with water, and lack sequence patterns. We tested the ability of E and K to resist nonspecific interactions through the design of a stealth material. Stealth materials are an ideal experimental system and provide a direct measure of nonspecific interactions. When a stealth material is coated onto surfaces, the degree of its resistance to nonspecific adsorption can be quantitatively measured using highly sensitive surface plasmon resonance (SPR) sensors. The choice of using both K and E is to balance charges and avoid a cationic or anionic surface. A random mix of E and K was used to test the conclusion that nature distributes E and K without a sequence pattern (Fig. 3a). Among the uncharged residues, N had the most similar properties to E and K, especially in its preference for protein surfaces (Fig. 3c).Experimental studies of protein adsorption on a peptide surface containing random K and E motifs were performed. The chosen sequence is Ac-[EK]7PPPPC-Am, where the square brackets indicate seven random E and K. Ac and Am indicate acetylation and amidation, respectively. The proline repeat and cysteine provide a stable anchor for self-assembling peptides on gold surfaces.16 The peptides were self-assembled onto a gold surface and adsorbed fibrinogen and lysozyme measured via SPR. The results are shown in Fig. 4. Untreated gold and poly-glycine were used as controls. The EK surface performance is comparable to the ultra-low protein fouling standard of <5 ng cm−2 of fibrinogen.35 Additionally a AC-[N]7PPPPC-Am peptide was synthesized because N had the best modeling results among the uncharged amino acids. It also had results below the ultra-low fouling threshold. See Fig. S5† for all SPR sensograms and Fig. S6† for peptide characterization.
![Protein adsorption results as determined by SPR. Ac-[EK]7PPPPC-Am (EK), Ac-GGGGGGGPPPPC-Am (G), and Ac-CPPPPNNNNNNN-Am (N) sequences were self-assembled onto gold and protein solution was flowed over. Bound protein after buffer wash is shown in the bars. Untreated gold and poly-glycine are shown for reference. EK and N show similar stealth performance.](/image/article/2012/SC/c2sc21135a/c2sc21135a-f4.gif) | ||
| Fig. 4 Protein adsorption results as determined by SPR. Ac-[EK]7PPPPC-Am (EK), Ac-GGGGGGGPPPPC-Am (G), and Ac-CPPPPNNNNNNN-Am (N) sequences were self-assembled onto gold and protein solution was flowed over. Bound protein after buffer wash is shown in the bars. Untreated gold and poly-glycine are shown for reference. EK and N show similar stealth performance. | ||
4 Conclusions
A fundamental understanding of nonspecific interactions has been gained by examining protein surfaces via bioinformatics. It was observed that (a) K and E are the most abundant on the surfaces of proteins and molecular chaperones, two disparate systems, but with variations in the distribution of each amino acid; (b) the KE content increases from extracellular proteins (21%) to cytoplasm proteins (27.5%) to molecular chaperones (38%) and to thermophilic molecular chaperones (52%); (c) K and E are distributed randomly; (d) K and E have strong water-binding capabilities, but weak binding with surrounding amino acids; (e) the uncharged amino acids which preferred water to other interactions were also abundant, N and S. These amino acids are used by proteins and molecular chaperones to resist nonspecific interactions, as supported by evidence from experiments with the random K and E peptides from this work, zwitterionic poly-EK alternating peptides from our previous studies13,16 and poly-serine by others.14,15 Understanding and mimicking nature's resistance to nonspecific interactions is key to addressing emerging challenges in chemistry, especially in practical applications where complex environments can degrade materials and surface coatings. The techniques and conclusions here provide new insights and directions into the understanding, characterization and design of nonspecific interactions.Acknowledgements
This work was supported by the Office of Naval Research (N00014-10-1-0600) and the National Science Foundation (CBET-0854298). AKN was supported by a National Science Foundation Graduate Fellowship. We would like to thank Dr Bill Howe and Prof. Vladimir Minin for providing valuable assistance.Notes and references
- B. D. Ratner and S. J. Bryant, Biomaterials: where we have been and where we are going, Annu. Rev. Biomed. Eng., 2004, 6, 41–75 CrossRef CAS.
- T. Ben-Yedidia, Design of peptide and polypeptide vaccines, Curr. Opin. Biotechnol., 1997, 8, 442–448 CrossRef CAS.
- K. P. Heidi, G. Kiss, T. M. Lee, R. Blomberg, R. A. Chica, L. M. Thomas, D. Hilvert, K. N. Houk and S. L. Mayo, Iterative approach to computational enzyme design, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 3790–3795 CrossRef.
- D. Farrusseng, S. Aguado and C. Pinel, Metal–organic frameworks: opportunities for catalysis, Angew. Chem., Int. Ed., 2009, 48, 7502–7513 CrossRef CAS.
- S. Jiang and Z. Q. Cao, Ultralow-fouling, functionalizable, and hydrolyzable zwitterionic materials and their derivatives for biological applications, Adv. Mater., 2010, 22, 920–932 CrossRef CAS.
- T. Peters, Jr, Serum albumin, Adv. Protein Chem., 1985, 37, 161–245 CrossRef.
- F. Veronese and A. Mero, The impact of PEGylation on biological therapies, BioDrugs, 2008, 22, 315–329 CrossRef CAS.
- B. Van den Berg, R. J. Ellis and C. M. Dobson, Effects of macromolecular crowding on protein folding and aggregation, EMBO J., 1999, 18, 6927–6933 CrossRef CAS.
- E. Harlow and D. Lane, Using Antibodies A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, 1998 Search PubMed.
- L. A. Horwich, W. A. Fenton, E. Chapman and G. W. Farr, Two families of chaperonin: physiology and mechanism, Annu. Rev. Cell Dev. Biol., 2007, 23, 115–145 CrossRef.
- P. B. Sigler, Z. Xu, H. S. Rye, S. G. Burston, W. A. Fenton and A. L. Horwich, Structure and function in GroEL-mediated protein folding, Annu. Rev. Biochem., 1998, 67, 581–608 CrossRef CAS.
- A. L. Horwich, C. Adrian and W. A. Fenton, The GroEL/GroES cis cavity as a passive anti-aggregation device, FEBS Lett., 2009, 583, 2654–2662 CrossRef CAS.
- S. Chen, Z. Q. Cao and S. Jiang, Ultra-low fouling peptide surfaces derived from natural amino acids, Biomaterials, 2009, 30, 5892–5896 CrossRef CAS.
- R. B. Olivier, M. C. Christopher, N. P. Joelle and M. Jean-Franois, Peptide self-assembled monolayers for label-free and unamplified surface plasmon resonance biosensing in crude cell lysate, Anal. Chem., 2009, 81, 6779–6788 CrossRef.
- R. Chelmowski, S. D. Köster, A. Kerstan, A. Prekelt, C. Grunwald, T. Winkler, N. Metzler-Nolte, A. Terfort and C. Wöll, Peptide-based SAMs that resist the adsorption of proteins, J. Am. Chem. Soc., 2008, 130, 14952–14953 CrossRef CAS.
- A. K. Nowinski, F. Sun, A. D. White, K. J. Keefe and S. Jiang, Sequence, structure, and function of peptide self-assembled monolayers, J. Am. Chem. Soc., 2012, 134, 6000–6005 CrossRef CAS.
- M. Ashburner, et al., Gene ontology: tool for the unification of biology, Nat. Genet., 2000, 25, 25–29 CrossRef CAS.
- C. Chaudhry, A. L. Horwich, A. T. Brunger and P. D. Adams, Exploring the structural dynamics of the E. coli chaperonin GroEL using translation-libration-screw crystallographic refinement of intermediate states, J. Mol. Biol., 2004, 342, 229–245 CrossRef CAS.
- T. Shimamura, A. Koike-Takeshita, K. Yokoyama, R. Masui, N. Murai, M. Yoshida, H. Taguchi and S. Iwata, Crystal structure of the native chaperonin complex from Thermus thermophilus revealed unexpected asymmetry at the cis-cavity, Structure, 2004, 12, 1471–1480 CrossRef CAS.
- J. H. Pereira, C. Y. Ralston, N. R. Douglas, D. Meyer, K. M. Knee, D. R. Goulet, J. A. King, J. Frydman and P. D. Adams, Crystal structures of a group II chaperonin reveal the open and closed states associated with the protein folding cycle, J. Biol. Chem., 2010, 285, 27958–27966 CrossRef CAS.
- M. M. U. Ali, S. M. Roe, C. K. Vaughan, P. Meyer, B. Panaretou, P. W. Piper, C. Prodromou and L. H. Pearl, Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex, Nature, 2006, 440, 1013–1017 CrossRef CAS.
- C. Dekker, S. M. Roe, E. A. McCormack, F. Beuron, L. H. Pearl and K. R. Willison, The crystal structure of yeast CCT reveals intrinsic asymmetry of eukaryotic cytosolic chaperonins, EMBO J., 2011, 30, 3078–3090 CrossRef CAS.
- R Development Core Team R, A Language and Environment for Statistical Computing, Vienna, Austria, http://www.R-project.org, 2008 Search PubMed.
- B. Howe, et al., Database-as-a-Service for Long Tail Science, SSDBM ’11: Proceedings of the 23rd Scientific and Statistical Database Management Conference, 2011 Search PubMed.
- N. R. Voss and M. Gerstein, Calculation of standard atomic volumes for RNA and comparison with proteins: RNA is packed more tightly, J. Mol. Biol., 2005, 346, 477–492 CrossRef CAS.
- J. Homola, Surface plasmon resonance sensors for detection of chemical and biological species, Chem. Rev., 2008, 108, 462–493 CrossRef CAS.
- W. Yang, H. Xue, W. Li, J. Zhang and S. Jiang, Pursuing “zero” protein adsorption of poly(carboxybetaine) from undiluted blood serum and plasma, Langmuir, 2008, 80, 7894–7901 Search PubMed.
- J. Homola, Surface Plasmon Resonance Based Sensors, Springer-Verlag, 2006 Search PubMed.
- R. Wolfenden, L. Andersson, P. M. Cullis and C. C. B. Southgate, Affinities of amino acid side chains for solvent water, Biochemistry, 1981, 20, 849–855 CrossRef CAS.
- M. A. Andrade, S. I. O'Donoghue and B. Rost, Adaptation of protein surfaces to subcellular location, J. Mol. Biol., 1998, 276, 517–525 CrossRef CAS.
- J. L. Jimenez, Does structural and chemical divergence play a role in precluding undesirable protein interactions, Proteins: Struct., Funct., Bioinf., 2005, 59, 757–764 CrossRef CAS.
- F. Glaser, D. M. Steinberg, I. A. Vakser and N. Ben-Tal, Residue frequencies and pairing preferences at protein–protein interfaces, Proteins: Struct., Funct., Genet., 2001, 43, 89–102 CrossRef CAS.
- P. Harder, M. Grunze, R. Dahint, G. M. Whitesides and P. E. Laibinis, Molecular conformation in oligo(ethylene glycol)-terminated self-assembled monolayers on gold and silver surfaces determines their ability to resist protein adsorption, J. Phys. Chem. B, 1998, 102, 426–436 CrossRef CAS.
- S. F. Chen, J. Zheng, L. Y. Li and S. Jiang, Strong resistance of phosphorylcholine self-assembled monolayers to protein adsorption: insights into nonfouling properties of zwitterionic materials, J. Am. Chem. Soc., 2005, 127, 14473–14478 CrossRef CAS.
- W. B. Tsia, J. M. Grunkemeir and T. A. Horbett, Human plasma fibrinogen adsorption and platelet adhesion to polystyrene, J. Biomed. Mater. Res., 1999, 44, 130–139 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional information on the sensitivity to the surface cutoff, details on the method of identifying interior surface residues, the glycine–serine sequence pair, the definition of interaction energy, a description of the calculation of error bars for each figure, and SPR sensograms. See DOI: 10.1039/c2sc21135a |
| This journal is © The Royal Society of Chemistry 2012 |