Structural insights into the potential of 4-fluoroproline to modulate biophysical properties of proteins†
Bastian
Holzberger
a,
Samra
Obeid
a,
Wolfram
Welte
b,
Kay
Diederichs
b and
Andreas
Marx
*a
aDepartment of Chemistry, Konstanz Research School Chemical Biology, University of Konstanz, Universitätsstr. 10, 78457, Konstanz, Germany. E-mail: Andreas.Marx@uni-konstanz.de; Fax: +49 7531 88 5140; Tel: +49 7531 88 5139
bDepartment of Biology, Konstanz Research School Chemical Biology, University of Konstanz, Universitätsstr. 10, 78457, Konstanz, Germany.
First published on 13th June 2012
Abstract
The unnatural amino acid 4-fluoroproline (4-FPro) can be used to replace natural proline in peptides and proteins to alter their stability, conformation and folding behavior. Interestingly, the two diastereomers (4R)- and (4S)-FPro behave quite differently resulting for example in increased or decreased protein stabilities. The reasons for the observed, opposed properties seem to be very complex and are not well understood yet, especially as only one single X-ray structure of a 4-FPro-modified protein is available, so far. The crystal structure of the large fragment of Taq DNA polymerase reported here far exceeds the molecular mass and number of 4-FPro residues of previous studied proteins and sheds light on how 4-FPro influences complex protein frameworks. It turns out that all aspects of prolyl Cγ-fluorination have to be considered in a combined fashion to understand how they account for the induced differences in protein stability. The interplay of different effects based on newly formed interactions and on the conformational preferences of 4-FPro determines whether the accepted diastereomer stabilizes or destabilizes the target protein. Due to counterbalanced effects, 4-FPro seems to be a very promising tool to even modify the properties of large enzymes with a high number of Pro residues since mono-fluorination at multiple sites is well tolerated by the target protein. Notably, the replacement of Pro by 4-FPro apparently also led to an improved crystallization capability of the DNA polymerase.
Introduction
Natural proline (Pro) is unique under the canonical amino acids as it displays a side chain that is covalently connected to its backbone nitrogen, forming a five-membered ring. Due to the resulting conformational restrictions Pro is unable to adopt most of the possible main chain conformations and is mainly located in loops, turns and at the start of helices.1,2 The prolyl ring prefers two puckering conformations that are defined as exo or endo with respect to the large out of plane displacement of the Cγ atom relative to the carbonyl group.3 Furthermore, Xaa-Pro peptide bonds exist in distinct cis and trans conformations whose isomerization plays a key role in protein folding.4 Thus, Pro influences protein conformations and the kinetics of protein folding.4–6 As there is no natural counterpart that might be used to replace Pro without drastic alterations, researchers focused on noncanonical analogs to modify peptides and proteins.7–204-Fluoroproline (4-FPro) has been shown to be very powerful in modulating stability, conformation and folding behavior of peptides and small model proteins.8–20 As the two diastereomers, (4R)- and (4S)-FPro, are known to directly influence the ratio of exo and endo puckering as well as the conformation of the prolyl peptide bond by stereoelectronic effects,8–10 efforts have been undertaken to use this as pre-organization effect for peptide engineering.11 Several small peptides were investigated concerning conformation, stability, and folding.12–15 It turned out that the two diastereomers (4R)- and (4S)-FPro can have antagonistic effects based on their bias to form either an exo puckering and a trans peptide conformation ((4R)-FPro) or to favor the endo puckering along with a cis peptide conformation ((4S)-FPro). These studies have rarely been expanded to globular proteins so far.16–24 Nevertheless, the two diastereomers also seem to behave differently in model proteins resulting either in enhanced or decreased stabilities as reported for Trp cage miniprotein (2.2 kDa),16 HP36 (4 kDa),17 and a barstar triple mutant (10 kDa).18,19 In each case one single Pro residue was replaced by either (4R)- or (4S)-FPro. Recently, three Pro residues in the model protein ubiquitin (8.6 kDa) were replaced by 4-FPro using recombinant protein expression in E. coli. Only (4R)-FPro yielded a protein with increased stability and slightly enhanced folding rates.20 In contrast, (4S)-FPro did not lead to any detectable protein expression in E. coli. Although the acceptance of either (4R)- or (4S)-FPro might be dictated by single sequence positions, the tendency that only one of the two diastereomers results in a properly folded protein was also reported for proteins harboring multiple Pro residues such as scFv (29 kDa) displaying eight Pro residues21 and EGFP (28 kDa) with ten Pro moieties.22 In both cases one diastereomer was well accepted leading to enhanced stability and accelerated folding whereas the other diastereomer resulted in insoluble, most probably unfolded proteins. These studies also show that both diastereomers can be processed by E. coli cells allowing in principle the expression of (4R)- and (4S)-FPro-modified target proteins. Taking into account that individual 4-FPro residues may contribute differently to altered properties, several reasons for the above mentioned results were discussed. Along with pre-organization forcing Pro residues to a certain puckering, the stabilization of the native peptide bond conformation was also mentioned.16–24 As the introduction of fluorine also leads to the formation of new interactions a variety of effects seems to influence proteins in a complex manner that is not well understood yet and has to be further addressed in detailed functional and structural studies. Unfortunately, only in the case of EGFP, a high-resolution crystal structure of a globular protein harboring 4-FPro moieties has so far been reported.22 Thus, it still remains speculative if the intrinsic conformational preferences of the 4-FPro diastereomers that have been mainly determined by studying N-acetylproline methyl esters8–10 actually take place in the complex framework of globular proteins. Furthermore, it has to be investigated how these preferences and the formation of new intramolecular non-covalent interactions account for altered biophysical properties of multi-fluorinated proteins.
Here, we present crystal structures of the large fragment of Taq DNA polymerase (KlenTaq, 61 kDa) in complex with its substrates. KlenTaq, harboring in total 32 Pro residues, is a flexible enzyme that undergoes several conformational transitions during catalysis. It exceeds the molecular mass and number of 4-FPro residues of previous 4-FPro-modified proteins by far. Recently, we have reported on the incorporation of 4-FPro in this enzyme and noticed that once more only one diastereomer, in this case (4R)-FPro, enabled the expression of a soluble and active protein whereas (4S)-FPro did not lead to protein expression in E. coli. Nevertheless, (4R)-FPro-KlenTaq retained enzymatic activity, fidelity, and sensitivity but displayed a loss of thermostability.25 To gain detailed insight into the structure of the multi-fluorinated (4R)-FPro-KlenTaq DNA polymerase for investigating the structural basis for the differences in stability along with the unaltered enzymatic properties and to further understand the opposed behavior of the two diastereomers, we characterized wild-type and fluorinated KlenTaq by CD spectroscopy and X-ray crystallography. The crystal structures of KlenTaq bearing either (4R)-FPro or natural Pro support the assumption that indeed the native prolyl peptide conformation determines the acceptance of only one 4-FPro diastereomer allowing protein expression and correct folding of the DNA polymerase in E. coli.
Results and discussion
Protein expression and characterization
KlenTaq DNA polymerase displays 32 Pro residues that are distributed over the whole protein sequence mainly located in loops, turns and random coils (Fig. 1). Furthermore, Pro is found as helix initiator and at bends of helices (Pro752 and Pro770). For the structural studies presented herein we expressed and purified wild-type and (4R)-FPro-KlenTaq without N-terminal tag that is known to hamper protein crystallization. The incorporation of (4R)-FPro was achieved by selective pressure incorporation (SPI) as previously described.25 Wild-type KlenTaq was expressed in standard LB medium. Afterwards, both enzymes were purified by anion exchange and size exclusion chromatography. For experimental details see the ESI.† | ||
| Fig. 1 Secondary structure of KlenTaq (PDB ID 3KTQ). Helices are shown in blue, strands are depicted in green, and Pro residues are highlighted with red dots. The residue numbering of full-length Taq DNA polymerase was used. | ||
To prove the replacement of Pro by (4R)-FPro we performed electrospray-ionization mass spectrometry. Interestingly, the N-terminal Met residue was already cleaved off at both the fluorinated and the wild-type enzyme. In case of (4R)-FPro-KlenTaq we observed two peaks that can be attributed to protein species with either all 32 Pro residues replaced by (4R)-FPro or with on average 31 residues substituted by (4R)-FPro (see Fig. S1 in the ESI†). This reflects a replacement level of more than 98%.
To study the differences of wild-type and (4R)-FPro-KlenTaq in terms of structure and thermostability we performed circular dichroism (CD) spectroscopy. Thereby, (4R)-FPro-KlenTaq exhibits a CD spectrum similar to the wild-type representing a largely unaltered overall fold (Fig. 2a). In accordance to literature local minima are observed at 208 and 222 nm representing the high α-helix content.26 Thermal denaturation was examined following the ellipticity at both minima (Fig. 2b). Considering the known dependence of KlenTaq's melting temperature on pH and scan rate the melting temperature of 101.8 °C for wild-type KlenTaq is in good accordance to literature.26 Interestingly, the melting temperature of (4R)-FPro-KlenTaq was only little lower (96.2 °C). Thus, the recently reported large difference in time-dependent assays at 95 °C between wild-type and (4R)-FPro-KlenTaq becomes understandable. There, it has been observed that wild-type KlenTaq is capable of retaining activity more than 5 h longer than (4R)-FPro-KlenTaq that shows already after 90 min at 95 °C less than 50% activity in primer extension reactions.25
 | ||
| Fig. 2 CD spectroscopy. a) CD spectra of wild-type (black) and (4R)-FPro-KlenTaq (red) in PBS buffer. b) Thermal denaturation of wild-type (black) and (4R)-FPro-KlenTaq (red) following the ellipticity at 208 (dots) and 222 nm (crosses) in PBS buffer. Depicted melting temperatures were determined by fitting two separate experiments (see Fig. S2 in the ESI†). | ||
Crystallization and structure determination
(4R)-FPro-KlenTaq was crystallized in complex with a DNA primer/template duplex and an incoming nucleoside triphosphate bound to the active site. We used 2′,3′-dideoxycytidine-5′-triphosphate (ddCTP) as a chain terminator leading to a stalled DNA polymerase due to the lack of the 3′-terminal hydroxyl group that is crucial for further primer elongation. Significant structural data of KlenTaq in ternary complexes have been already created by this method27–29 including the respective structure in complex with DNA and ddCTP solved at a resolution of 2.3 Å (PDB ID 3KTQ).27 Nevertheless, for a direct comparison, we also crystallized wild-type KlenTaq to take advantage of the improvement of refinement programs over the last few years. We observed that fluorinated KlenTaq crystallized under a broader range of crystallization conditions and faster than the wild-type protein. Possible reasons for the improved crystallization behavior upon (4R)-FPro introduction are summarized and discussed later. Crystallographic data were collected, the structures were solved by difference Fourier techniques, and refined at a resolution of 2.4 Å in case of (4R)-FPro-KlenTaq and 1.9 Å for the wild-type protein (see Table S1 in the ESI†).Comparison of the overall structures
(4R)-FPro-KlenTaq exhibits a structure that is very similar to the wild-type enzyme resulting in a root mean square deviation for Cα atoms of only 0.65 Å (Fig. 3a). Although both structures are closely related to the known ternary complex (PDB ID 3KTQ)27 we found some differences. In accordance with other recently published ternary complexes of KlenTaq DNA polymerase30–32 we found cis peptide bonds preceding Pro300 and Pro579 (Fig. 3) instead of the trans conformations as found in PDB IDs 3KTQ,271QSS, 1QTM, and 1QSY.28 Furthermore, our high-resolution wild-type structure suggests a second conformation of Glu786 that might contribute to magnesium ion coordination in the active site (see Fig. S3 in the ESI†). This conformation has been proposed only in PDB ID 3PO5,32 so far. | ||
| Fig. 3 Structures of wild-type (grey) and (4R)-FPro-KlenTaq (red) in ternary complex with a DNA primer/template complex and ddCTP bound in the active site. Pro and (4R)-FPro residues are highlighted with black and red sticks, respectively. The structure elements fingers, thumb and palm are labeled. a) Superimposed structures. b) Close-up view of wild-type KlenTaq around Pro300. c) Close-up view of (4R)-FPro-KlenTaq around (4R)-FPro579. The refined models are shown in 2mFo-DFc maps at 1σ. | ||
Most of the parts harboring Pro and (4R)-FPro residues are very well resolved allowing reliable determination of the conformations and their surroundings (e.g. see Fig. 3b, c). Although the introduction of fluorine leads to alterations in prolyl ring puckering and the formation of new interactions as discussed later, there are no major structural alterations of the overall protein fold in comparison to the wild-type enzyme. Thus, the unaltered enzymatic properties of (4R)-FPro-KlenTaq25 are first of all due to the structural similarities of wild-type and fluorinated enzyme.
Prolyl ring puckering
To obtain detailed insight into prolyl ring puckering (Fig. 4a), we calculated for wild-type and (4R)-FPro-KlenTaq in each case two simulated annealing omit maps, mFo-DFc, for either Cγ (and F) or Cβ, Cγ, Cδ (and F). Based on these omit maps the prolyl ring puckerings were determined. In a second step after refinement, the resulting conformations were visually verified and adjusted by model rebuilding. After further refinement steps, we calculated again simulated annealing omit maps for the respective prolyl side chain atoms to confirm the final conformations (Fig. 4b, c). Advantageously, the wild-type structure is resolved to 1.9 Å and thus allowed the reliable determination of the prolyl puckering in 28 out of 32 cases. Although the structure of (4R)-FPro-KlenTaq is only resolved to 2.4 Å the additional fluorine atoms facilitate the assignment of puckerings to the (4R)-FPro residues and allowed also the determination of 28 (4R)-FPro conformations. For 18 Pro and 26 (4R)-FPro residues one distinct conformation was clearly defined whereas for the remaining 10 and two moieties, respectively, two conformations were used to properly fit the model to the respective electron densities. Unfortunately, due to the lack of sufficient electron density, in particular in some parts of the flexible fingers domain, we were not able to reliably assign prolyl ring conformations to Pro650, 656, 685, and 701 as well as (4R)-FPro336, 527, 650, and 701 (Fig. 4b, c). An overview of all values determined for Pro and (4R)-FPro residues, is shown in the ESI Tables S2 and S3.†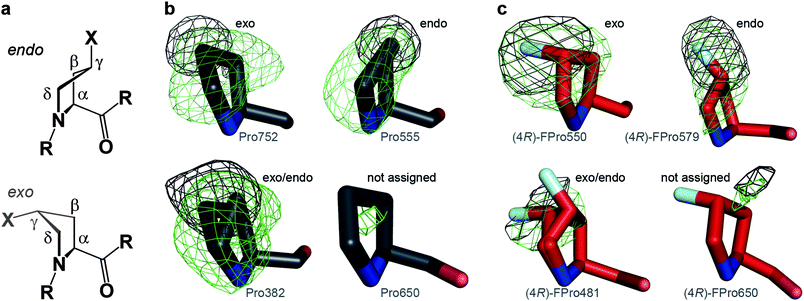 | ||
| Fig. 4 Determination of prolyl ring puckering applying simulated annealing omit maps, mFo-DFc, at 3σ for all side chain atoms (green) or only Cγ (and F) (black). a) Endo and exo puckering of (4R)-FPro (X = F) and Pro (X = H), respectively. The four atoms Cα, Cβ, Cδ and N constitute one plane and Cγ is puckered out of plane (envelope). b) Exemplary Pro residues and c) exemplary (4R)-FPro residues displaying exo or endo puckerings or two alternative conformations. Pro650 and (4R)-FPro650 were not considered for further interpretations as sufficient electron density is missing. | ||
The analysis of backbone torsion angles (Fig. 5a) of Pro and (4R)-FPro residues resulted in values clustered around Φ,Ψ = −60°, −30° and Φ,Ψ = −75°, +150° (Fig. 5b). The prolyl ring typically prefers two puckering conformations that are defined as exo or endo with respect to the large out of plane displacement of the Cγ atom relative to the carbonyl group. Although in most cases envelope conformations are depicted for simplicity (Fig. 4a) Pro would rather adopt twisted conformations with mainly Cβ and Cγ out of plane.3,33 The relative position of Cγ is determined by the side chain torsion angle χ1 displaying negative values for an exo puckering and positive angles for an endo conformation3 or, alternatively, by considering all four side chain torsion angles (χPro = χ1 + χ3 − χ2 − χ4) exhibiting either χPro > +40° (endo) or < −40° (exo).34 For wild-type and (4R)-FPro-KlenTaq it turned out that χ1 values (Fig. 5c) assemble in both structures either at χ1 ≈ −25° (exo) or > +10° (endo). The respective criterion for χPro is also fulfilled for all investigated Pro and (4R)-FPro residues except for the second conformation of (4R)-FPro300 that seems to be endo related but shows for χ1 a slightly negative value (−1.7°). This is caused by an exceptional envelope conformation with Cδ out of plane.
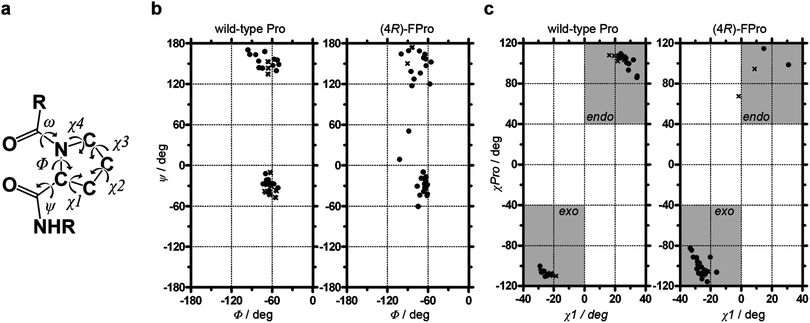 | ||
| Fig. 5 Torsion angle analysis of Pro and (4R)-FPro residues. a) Definition of torsion angles. b) Ramachandran plots for all Pro and (4R)-FPro residues of wild-type (left) and (4R)-FPro-KlenTaq (right). c) Assignment of Cγ prolyl puckering considering χ1 and χPro (χ1 + χ3 − χ2 − χ4). Pro650, 656, 685, and 701 as well as (4R)-FPro336, 527, 650, and 701 are not shown in (c) as no reliable conformation could be assigned. Residues with one single conformation are shown with dots; residues displaying two possibilities are depicted with one cross each. | ||
(4R)-FPro residues show predominantly an exo puckering (89%) whereas only 43% of native Pro residues are exo, but 57% show an endo related puckering (alternative conformations were counted with 0.5 each). Although the exact and reliable assignment of prolyl ring puckering by X-ray crystallography may be not far beyond the limits of detection, this unambiguously shows the tendency of (4R)-FPro to adopt exo conformations even at different positions in a large globular protein. In absolute terms, six residues are natively pre-organized in their exo conformation but seven Pro residues are switched from an endo conformation to an exo-puckering due to the introduction of (4R)-FPro (see Tables S2 and S3 in the ESI†). Furthermore, four natively endo puckered residues do not adopt the favored exo puckering in (4R)-FPro-KlenTaq, rather, they remain in the less favored endo, or show both conformations. The remaining nine investigated Pro residues do not show any bias for exo or endo puckering in the native state but are completely shifted to the exo conformation in (4R)-FPro-KlenTaq. Thus, the introduction of (4R)-FPro switches more prolyl rings to an exo conformation than pre-organizing Pro residues in their native conformation. This may account for the observed loss of stability but does not explain why (4S)-FPro does not lead to a properly folded protein since native Pro residues display even slightly more endo conformations than exo-puckered prolyl rings and (4S)-FPro should have been favored in this aspect.
Notably, in wild-type KlenTaq 36% (10 out of 28) of the well-defined Pro residues show two prolyl puckering conformations whereas in (4R)-FPro-KlenTaq only 2 out of 28 analyzed residues (7%) display two conformations. The Cγ-fluorination thus removes ambiguities in conformation by favoring one conformation over the other. The resulting reduced conformational heterogeneity may account for the improved crystallization properties of (4R)-FPro-KlenTaq.
Conformation of Xaa-Pro peptide bonds
Natural Pro is known to show the cis conformation (ω = 0°) in 5–6% of preceding peptide bonds whereas all other canonical amino acids almost exclusively display trans peptide bonds (ω = 180°).5,35 Correspondingly, wild-type KlenTaq reveals two cis-Pro residues (Pro300 and 579). Although previous X-ray structures of KlenTaq DNA polymerase largely confirm this assignment we have verified the two cis peptide bonds that are clearly defined by model maps (Fig. 3b, c) by refining trans conformations as a validation step. Doing so, the existence of cis conformations was confirmed as no reliable electron densities were detectable for trans peptide bonds. Interestingly, the two cis-Pro residues are endo-puckered (see Table S2 in the ESI†) in accordance to an analysis of high-resolution X-ray structures indicating that 89% of cis-Pro residues show an endo conformation.34Although (4R)-FPro is known to stabilize a trans peptide bond, we found cis conformations for these two residues also in (4R)-FPro-KlenTaq. This suggests that Cγ-fluorination is not capable of switching peptide bond conformations in KlenTaq DNA polymerase since this would require major local rearrangements. Interestingly, (4R)-FPro579 retains the less favored endo puckering whereas (4R)-FPro300 shows two conformations, the favored exo puckering and an endo related envelope conformation with Cδ out of plane. Hence, two of the four residues ((4R)-FPro300, 301, 481, and 579) that do not exclusively adopt exo puckerings are preceded by cis peptide bonds. As both cis-Pro moieties show natively an endo conformation the respective cis-(4R)-FPro residues seem to avoid a pure exo ring puckering. Apparently, the interdependency of prolyl ring puckering and cis/trans property of preceding peptide bonds that has been observed for Cγ-fluorinated N-acetylproline methyl esters8 also takes place in globular proteins and hampers exo puckering for cis-(4R)-FPro.
Interactions and surface exposure of fluorine
Pro does not take part in typical noncovalent interactions within globular proteins such as hydrogen bonds, cation-π, major electrostatic or hydrophobic contacts. In contrast, natural Pro residues are nowadays discussed to be involved in nonconventional interactions to aromatic- and carbonyl-groups.36–38 By analyzing PDB files, 45% of Pro moieties have been found to form nonconventional hydrogen bonds to aromatic side chains and 74% have even been shown to undergo interactions with neighboring amide carbonyls (36% displayed both interactions). Thereby, Cα and Cδ of the prolyl ring were thought to act as hydrogen donors as they are acidified by the adjacent carbonyl group and the nitrogen atom, respectively.36In KlenTaq DNA polymerase most of the Pro residues are in contact with neighboring amino acids displaying Cδ hydrogens that interact with adjacent amide carbonyls or aromatic rings (e.g. see Fig. 6a, b). The introduction of the highly electronegative fluorine atom at Cγ leads to further acidification of adjacent ring hydrogens and thus may strengthen such hydrogen bonds.17 In (4R)-FPro-KlenTaq such interactions of Cγ hydrogens to e.g. an aromatic ring can be also detected (Fig. 6c). However, the enhanced steric demand of fluorine compared to hydrogen39 and the intrinsic bias of (4R)-FPro to adopt an exo puckering may alter preferred conformations and might not lead to more favorable interactions at all sites in comparison to the natural case.
 | ||
| Fig. 6 Local microenvironments of exemplary Pro and (4R)-FPro residues in wild-type (grey) and (4R)-FPro-KlenTaq (red). Fluorine is shown in cyan and interactions are either green or red (repulsions). Distances are given in Å. a) Pro752 interacts with adjacent amide carbonyls via Cδ-H⋯O bridges. b) Pro585 next to the 3′-end of the DNA primer strand: Cδ hydrogens interact with a tyrosine side chain via aromatic-Pro bridges. c) (4R)-FPro585: Cδ hydrogens interact with a tyrosine side chain via aromatic-Pro bridges, fluorine is in van der Waals contact to a leucine residue and is apparently involved in a repulsive interaction with the phosphate of the DNA primer strand. d) (4R)-FPro752 interacts with a valine side chain via van der Waals contacts, with an adjacent amide bonds via dipole interactions and shows a repulsive interaction to an amide carbonyl. e) (4R)-FPro501 displays a fluorine atom in close proximity to a repulsive carbonyl oxygen. f) (4R)-FPro579's fluorine atom points towards a hydrophobic patch forming several van der Waals contacts. | ||
Furthermore, the introduction of fluorine adds to the interplay of noncovalent interactions as additional interactions are formed in the microenvironments of (4R)-FPro residues (Fig. 6c–f). Thus, in (4R)-FPro-KlenTaq not only dipole–dipole interactions between the highly polarized C–F bond and adjacent polar groups are represented at multiple sites (e.g. see Fig. 6d, e) but also new van der Waals contacts can be found (Fig. 6f). Amongst them, interactions to the DNA substrate in close proximity to the 3′-end of the DNA primer are present as well (Fig. 6c). Notably, (4R)-FPro501 seems to accept a repulsive interaction to the carbonyl oxygen rather than flipping its ring conformation to an intrinsically less favored endo conformation that would avoid this repulsion (Fig. 6e), confirming the preference of (4R)-FPro for an exo-puckered ring even in the face of such repulsive interactions.
To estimate the total number of newly formed interactions in (4R)-FPro-KlenTaq, we counted contacts of the 32 fluorine atoms to all atoms that are less than 4.5 Å apart and are at least partially charged or part of dipoles. We detected interactions to carbons, nitrogens and oxygens of amide bonds, to acidic and hydrophilic side chains, to nitrogens in tryptophan and histidine side chains or to hydroxyl groups of serine and tyrosine. In total more than 100 interactions of fluorine atoms to adjacent polar groups are possible albeit van der Waals contacts were not included. Although a ranking or weighting of the single contacts is not reasonable, no notable tendency for favored or repulsive interactions was detected. Taken together, a major contribution to modified protein stabilities can be expected from this newly formed network of favorable and repulsive interactions. Thus, along with the estimated alterations of already existent interactions of native Pro residues the stability of KlenTaq DNA polymerase is altered in a highly counterbalanced fashion. Unfortunately, the high number of fluorinated Pro moieties amplifies the complexity of the system and hampers predictions on protein stability even with detailed structural information. However, we do not observe any rearrangements of amino acid side chains or substrates reflecting that Cγ-fluorination of Pro residues is well tolerated without disturbing the native structure - although fluorine is not only different in electronegativity but also in size.39 In this regard, the accommodation of (4S)-FPro by KlenTaq DNA polymerase can not be excluded by its different stereochemistry or the preference for an endo puckering albeit it may lead to varied consequences on protein stability due to a different network of interactions.
Crystallization behavior
Aside from interacting with adjacent amino acids as discussed above, (4R)-FPro residues also change the properties of KlenTaq's surface. As only about one third of the 32 (4R)-FPro residues are buried within the protein, the majority of fluorine atoms in (4R)-FPro-KlenTaq are exposed to the solvent forming a fluorinated surface (Fig. 7). | ||
| Fig. 7 Surface representation of (4R)-FPro-KlenTaq. The protein is shown in red. DNA template is depicted in orange, the primer is yellow. Fluorine atoms are cyan. | ||
Notably, six of these exposed (4R)-FPro residues, namely (4R)-FPro298, 336, 481, 685, 812, and 816 even display fluorine atoms that are in close contact to symmetry-related KlenTaq molecules and are thus, capable of forming new crystallization contacts. Accompanied by the heavily fluorinated surface these interactions may in sum account for the improved crystallization behavior of (4R)-FPro-KlenTaq in comparison to the wild-type enzyme. Aside from the multi-fluorinated protein surface the reduced local heterogeneities may additionally contribute to the enhanced crystallization competence of (4R)-FPro-KlenTaq. Interestingly, facilitated crystal formation has also been reported for the 4-FPro modified EGFP even though Budisa and coworkers ascribed this effect neither to decreased conformational heterogeneity or a fluorinated surface nor to new crystal contacts, but to an increased hydrophobicity of buried Pro residues and the resulting stabilization of the fluorinated protein.22
Conclusion
The global replacement of Pro by (4R)-FPro neither influences neighboring side chain conformations nor the overall structure of KlenTaq, which explains the reported similar enzymatic properties of (4R)-FPro-KlenTaq.25 Thus, (4R)-FPro does not markedly disrupt protein structures and is therefore also capable of maintaining enzymatic activities. However, differences in the stability of target proteins due to 4-FPro incorporation are most likely based on a counterbalanced interplay of several effects: the establishment of a huge network of new and different noncovalent interactions introduced by Cγ-fluorination of Pro residues, the acceptance of fluorine atoms in either buried or exposed positions and the tendency of e.g. (4R)-FPro to switch prolyl ring puckerings into an exo conformation, regardless of their native form. Accordingly, in fluorinated KlenTaq most of the prolyl rings adopt exclusively the exo conformation reducing structural heterogeneity.As KlenTaq DNA polymerase is the first protein that shows a decreased stability when incorporating one of the two 4-FPro diastereomers, but is not successfully expressed in presence of the other one, we suggest that acceptance and influence on protein stability are not compulsorily interrelated. Rather, the acceptance of one diastereomer during protein expression in vivo seems to rely on effects different from final protein stability. Thereby, differences resulting in altered protein solubility or modified folding pathways presumably associated with cis–trans isomerization of Pro peptide bonds may play important roles. At least for KlenTaq DNA polymerase, the existence of 30 trans- but only two cis-Pro peptide bonds might have been the knock-out criterion for the expression of KlenTaq DNA polymerase in presence of (4S)-FPro that stabilizes the cis conformation. On the other hand, EGFP, which adopts the trans conformation in nine out of ten Pro residues, can be expressed in presence of (4S)-FPro whereas (4R)-FPro results in an unfolded and insoluble protein.22 Nevertheless, further in vivo expression experiments on proteins that display almost exclusively trans-Pro peptide bonds20,21 have also reported on successful protein expression only in presence of (4R)-FPro. Thus, at least for KlenTaq DNA polymerase the most probable reason for prevented target protein expression in presence of (4S)-FPro seems to be the predominance of trans-Pro residues even though this might not be a universal criterion.
Notably, the replacement of Pro by 4-FPro apparently also led to an improved crystallization capability of KlenTaq DNA polymerase. This is most likely caused by the multi-fluorinated protein surface capable of forming new crystal contacts, reduced local heterogeneities, and modified hydrophobic potentials.
In summary, the overall effects of globally substituting Pro residues by (4R)-FPro are difficult to predict. However, 4-FPro is suitable for the engineering of large proteins and enzymes even with a high number of Pro residues as local and global structures are not markedly altered upon Cγ-fluorination of Pro residues. Thus, biophysical properties like folding, stability, or crystallization behavior can be modulated with 4-FPro.
Methods
CD spectroscopy
The buffer of KlenTaq samples was exchanged by 1 × PBS buffer (137 mM NaCl, 2.7 mM KCl, 10.2 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4) using Viva Spin columns (several dilution and re-concentration steps). CD spectra (250 μL, 1.5 μM KlenTaq, quartz cuvettes, light path 1 mm, 0.1 nm data intervals, 20 °C) were averaged from 6 scans. Denaturation curves were recorded following the ellipticity at 208 and 222 nm in dependence of the temperature (0.2 °C min−1). According to thermal denaturation studies reported in literature,26 we used a potassium phosphate based buffer, in our case PBS buffer (pH 7.4), and sealed the cuvettes with a stopper and parafilm laboratory film. As the applied 1 mm quartz cuvettes display a rather small remaining expansion space, evaporation effects were kept minimal. Data were collected at every 0.5 °C. Melting temperatures were determined by sigmoidal fitting applying a linear increasing baseline and calculating the derivatives (see Figure S2 in the ESI†). Unfortunately, thermal unfolding led to sample precipitation.Crystallization
Crystallization was conducted using a solution containing 105 μM KlenTaq, 16 mM Tris·HCl (pH 7.5), 120 mM NaCl, 0.8 mM EDTA, 0.8 mM β-mercaptoethanol, 158 μM DNA template (5′-d(AAA GGG CGC CGT GGT C)-3′), 158 μM DNA primer (5′-d(GAC CAC GGC GC)-3′), 1.1 mM ddCTP, 19.2 mM MgCl2 and mixed in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with 1 μL reservoir solution (wild-type KlenTaq: 50 mM sodium cacodylate (pH 6.5), 0.2 M ammonium acetate, 10 mM magnesium acetate, 15% (w/v) PEG 8000; (4R)-FPro-KlenTaq: 0.1 M HEPES (pH 7.5), 20 mM manganese chloride, 0.1 M ammonium acetate, 15% (w/v) PEG 4000). Crystals were produced by sitting drop vapor diffusion equilibrating against 100 μL of the reservoir solution. After two weeks at 18 °C crystals were frozen in liquid nitrogen.
:1 ratio with 1 μL reservoir solution (wild-type KlenTaq: 50 mM sodium cacodylate (pH 6.5), 0.2 M ammonium acetate, 10 mM magnesium acetate, 15% (w/v) PEG 8000; (4R)-FPro-KlenTaq: 0.1 M HEPES (pH 7.5), 20 mM manganese chloride, 0.1 M ammonium acetate, 15% (w/v) PEG 4000). Crystals were produced by sitting drop vapor diffusion equilibrating against 100 μL of the reservoir solution. After two weeks at 18 °C crystals were frozen in liquid nitrogen.
Data collection and structure determination
Datasets were collected from cryo-cooled crystals at the Swiss Light Source (Paul Scherrer Institute, Villingen, Switzerland) using the beamline PXIII (X06DA) at a wavelength of 1.23 Å (wild-type KlenTaq) and 1.00 Å ((4R)-FPro-KlenTaq) and a Mar225 CCD detector. Data reduction was performed with the XDS package40 and structures were solved by difference Fourier techniques using wild-type KlenTaq (PDB ID 3KTQ) as model. Refinement was performed with PHENIX41 and model rebuilding was done with COOT.42 The structures were solved and refined to a resolution of 1.9 Å (wild-type) and 2.4 Å ((4R)-FPro-KlenTaq), respectively. Both structures were in the same space group P3121, with cell dimensions a = b = 107.8, c = 89.6 Å (wild-type) and a = b = 109.6, c = 91.0 Å ((4R)-FPro-KlenTaq), respectively. The two bivalent ions in the active site of wild-type KlenTaq were Mg2+ whereas we found for (4R)-FPro-KlenTaq one Mg2+ and one Mn2+ ion due to crystallization conditions. Figures were made with PyMOL.43Coordinates and structure factors have been deposited in the Protein Data Bank with the accession numbers 4DLG (wild-type KlenTaq) and 4DLE ((4R)-FPro-KlenTaq).
Acknowledgements
Financial Support through the Konstanz Research School Chemical Biology and the DFG is gratefully acknowledged. B. H. thanks the Carl-Zeiss-Foundation for a Ph.D. scholarship. We also thank H.-P. Brachvogel for the analysis of the torsion angles, K. Bergen and K. Betz for the help with crystal structure data collection and analysis, and M. Rubini for helpful discussion. We gladly acknowledge beamtime and support at the Swiss Light Source (PSI, Villigen, Switzerland).Notes and references
- M. J. Betts and R. B. Russell, in Bioinformatics for Geneticists, ed. M. R. Barnes, Wiley-VCH, Weinheim, Germany, 2nd edn, 2007 Search PubMed.
- B. K. Kay, M. P. Williamson and M. Sudol, FASEB J., 2000, 14, 231–241 CAS.
- F. A. Momany, R. F. McGuire, A. W. Burgess and H. A. Scheraga, J. Phys. Chem., 1975, 79, 2361–2381 CrossRef CAS.
- W. J. Wedemeyer, E. Welker and H. A. Scheraga, Biochemistry, 2002, 41, 14637–14644 CrossRef CAS.
- K. P. Lu, G. Finn, T. H. Lee and L. K. Nicholson, Nat. Chem. Biol., 2007, 3, 619–629 CrossRef CAS.
- M. W. MacArthur and J. M. Thornton, J. Mol. Biol., 1991, 218, 397–412 CrossRef CAS.
- R. S. Erdmann and H. Wennemers, Angew. Chem., Int. Ed., 2011, 50, 6835–6838 CrossRef CAS.
- M. L. DeRider, S. J. Wilkens, M. J. Waddell, L. E. Bretscher, F. Weinhold, R. T. Raines and J. L. Markley, J. Am. Chem. Soc., 2002, 124, 2497–2505 CrossRef CAS.
- E. S. Eberhardt, N. Panisik Jr. and R. T. Raines, J. Am. Chem. Soc., 1996, 118, 12261–12266 CrossRef CAS.
- L. E. Bretscher, C. L. Jenkins, K. M. Taylor, M. L. DeRider and R. T. Raines, J. Am. Chem. Soc., 2001, 123, 777–778 CrossRef CAS.
- M. D. Shoulders, K. A. Satyshur, K. T. Forest and R. T. Raines, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 559–564 CrossRef CAS.
- M. D. Shoulders and R. T. Raines, Annu. Rev. Biochem., 2009, 78, 929–958 CrossRef CAS.
- Y.-C. Chiang, Y.-J. Lin and J.-C. Horng, Protein Sci., 2009, 18, 1967–1977 CrossRef CAS.
- J.-C. Horng and R. T. Raines, Protein Sci., 2006, 15, 74–83 CrossRef CAS.
- W. Kim, R. A. McMillan, J. P. Snyder and V. P. Conticello, J. Am. Chem. Soc., 2005, 127, 18121–18132 CrossRef CAS.
- D. Naduthambi and N. J. Zondlo, J. Am. Chem. Soc., 2006, 128, 12430–12431 CrossRef CAS.
- T.-Y. Zheng, Y.-J. Lin and J.-C. Horng, Biochemistry, 2010, 49, 4255–4263 CrossRef CAS.
- R. Golbik, C. Yu, E. Weyher-Stingl, R. Huber, L. Moroder, N. Budisa and C. Schiene-Fischer, Biochemistry, 2005, 44, 16026–16034 CrossRef CAS.
- C. Renner, S. Alefelder, J. H. Bae, N. Budisa, R. Huber and L. Moroder, Angew. Chem., Int. Ed., 2001, 40, 923–925 CrossRef CAS.
- M. D. Crespo and M. Rubini, PLoS One, 2011, 6, e19425 CAS.
- S. Edwardraja, S. Sriram, R. Govindan, N. Budisa and S.-G. Lee, Mol. BioSyst., 2011, 7, 258–265 RSC.
- T. Steiner, P. Hess, J. H. Bae, B. Wiltschi, L. Moroder and N. Budisa, PLoS One, 2008, 3, e1680 Search PubMed.
- L. Merkel, M. Schauer, G. Antranikian and N. Budisa, ChemBioChem, 2010, 11, 1505–1507 CrossRef CAS.
- S. Lepthien, L. Merkel and N. Budisa, Angew. Chem., Int. Ed., 2010, 49, 5446–5450 CrossRef CAS.
- B. Holzberger and A. Marx, J. Am. Chem. Soc., 2010, 132, 15708–15713 CrossRef CAS.
- I. Karantzeni, C. Ruiz, C.-C. Liu and V. J. LiCata, Biochem. J., 2003, 374, 785–792 CrossRef CAS.
- Y. Li, S. Korolev and G. Waksman, EMBO J., 1998, 17, 7514–7525 CrossRef CAS.
- Y. Li, V. Mitaxov and G. Waksman, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9491–9496 CrossRef CAS.
- Y. Li and G. Waksman, Protein Sci., 2001, 10, 1225–1233 CrossRef CAS.
- K. Betz, F. Streckenbach, A. Schnur, T. Exner, W. Welte, K. Diederichs and A. Marx, Angew. Chem., Int. Ed., 2010, 49, 5181–5184 CrossRef CAS.
- S. Obeid, A. Baccaro, W. Welte, K. Diederichs and A. Marx, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21327–21331 CrossRef CAS.
- S. Obeid, A. Schnur, C. Gloeckner, N. Blatter, W. Welte, K. Diederichs and A. Marx, ChemBioChem, 2011, 12, 1574–1580 CrossRef CAS.
- G. Némethy, K. D. Gibson, K. A. Palmer, C. N. Yoon, G. Paterlini, A. Zagari, S. Rumsey and H. A. Scheraga, J. Phys. Chem., 1992, 96, 6472–6484 CrossRef.
- E. J. Milner-White, L. H. Bell and P. H. Maccallum, J. Mol. Biol., 1992, 228, 725–734 CrossRef CAS.
- H. Reiersen and A. R. Rees, Trends Biochem. Sci., 2001, 26, 679–684 CrossRef CAS.
- R. Bhattacharyya and P. Chakrabarti, J. Mol. Biol., 2003, 331, 925–940 CrossRef CAS.
- T. Steiner and G. Koellner, J. Mol. Biol., 2001, 305, 535–557 CrossRef CAS.
- P. Chakrabarti and S. Chakrabarti, J. Mol. Biol., 1998, 284, 867–873 CrossRef CAS.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- W. Kabsch, J. Appl. Crystallogr., 1993, 26, 795–800 CrossRef CAS.
- P. D. Adams, R. W. Grosse-Kunstleve, L.-W. Hung, T. R. Ioerger, A. J. McCoy, N. W. Moriarty, R. J. Read, J. C. Sacchettini, N. K. Sauter and T. C. Terwilliger, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2002, 58, 1948–1954 CrossRef.
- P. Emsley and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef.
- W. L. DeLano, The PyMOL User's Manual, DeLano Scientific, Palo Alto, CA, USA, 2002 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Methods for mass spectrometry and protein expression and purification; SDS PAGE gel of purified wild-type and (4R)-FPro-KlenTaq; CD spectroscopy; Thermal denaturation curves; Active site of wild-type KlenTaq; Data collection and refinement statistics; Torsion angles, peptide bond conformations, and prolyl ring puckering conformations. See DOI: 10.1039/c2sc20545a |
| This journal is © The Royal Society of Chemistry 2012 |