 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Physical mixing of metal acetates: a simple, scalable method to produce active chloride free bimetallic catalysts†
Simon A.
Kondrat
a,
Greg
Shaw
a,
Simon J.
Freakley
a,
Qian
He
b,
Joanna
Hampton
a,
Jennifer K.
Edwards
a,
Peter J.
Miedziak
a,
Thomas E.
Davies
a,
Albert F.
Carley
a,
Stuart H.
Taylor
a,
Christopher. J.
Kiely
b and
Graham J.
Hutchings
*a
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, UK, CF10 3AT. E-mail: hutch@cardiff.ac.uk; Fax: +44 (0)29 2087 4059
bDepartment of Materials Science and Engineering, Lehigh University, 5 East Packer Avenue, Bethlehem, PA 18015-3195, USA
First published on 16th July 2012
Abstract
We have prepared supported gold, palladium and gold–palladium bimetallic catalysts by the physical mixing of the acetate salts of the metals followed by a simple heat treatment. The use of the acetates as the metal precursor eliminates chloride from the catalyst preparation step. Extensive characterisation shows the formation of bimetallic alloy particles. These catalysts are extremely active for alcohol oxidations and the direct formation of hydrogen peroxide.
Introduction
Heterogeneous catalysis is a key process in the manufacture of a large range of bulk and fine chemicals and lies at the heart of green chemical processes. The use of catalysts for oxidation reactions can lead to a greener process when compared with alternative routes. Gold nanoparticles have been shown to be effective for the oxidation of alkenes1–3 and alcohols,4,5 the oxidation of CO6,7 and for the direct synthesis of hydrogen peroxide.8 It has also been demonstrated that alloying gold with palladium leads to significant enhancement in activity for alcohol oxidation9,10 and also markedly improves the yield of hydrogen peroxide in the direct synthesis reaction.10,11The catalysts reported for these oxidations are usually prepared by three main methods; namely, wet impregnation, deposition precipitation and sol-immobilisation. However, all these preparation techniques use HAuCl4 as the gold precursor, making it difficult to completely remove chloride from the final catalyst. It is known that the presence of chloride in a catalyst can lead to particle agglomeration, through Au–Cl–Au bridges12 and also leads to the blocking of active sites.13 The result of these affects is a loss of catalyst activity, which is well documented for CO oxidation with gold monometallic catalysts.14,15 It has been observed that a Cl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Au atom ratio of 0.1 is sufficient to decrease the activity by half for a CO oxidation catalyst, despite most of the Cl− being associated solely with the Al2O3 support. The blocking of sites on the Al2O3 support with phosphate showed that a Cl:Au atom ratio even as low as 0.0006 could affect activity.
:Au atom ratio of 0.1 is sufficient to decrease the activity by half for a CO oxidation catalyst, despite most of the Cl− being associated solely with the Al2O3 support. The blocking of sites on the Al2O3 support with phosphate showed that a Cl:Au atom ratio even as low as 0.0006 could affect activity.
A method of limiting chloride levels in catalysts involves tuning the pH of the deposition, to ensure significant hydrolysis of the HAuCl4 and also to minimize Cl− deposition onto the support.15 However, it is known that a pH that facilitates total hydrolysis (∼pH 10) also results in a decrease in the Au loading levels.15,16 An alternative method of reducing chloride levels is by heat treatment of the catalyst, but unfortunately this causes agglomeration through sintering.15
Metal acetate shows significant advantages over other frequently used precursors as it is halide free, and clearly has potential for the chloride-free formation of Au and AuPd catalysts. Chloride-free Au supported on Al2O3, for selective CO oxidation, has previously been produced by the use of an aqueous suspension of the acetate followed by calcination.13 An alternative process has been shown to be effective for the formation of metal nanoparticles on carbon nanotube supports, by the simple physical mixing of acetate salts with the nanotubes and subsequent heat treatment under an inert atmosphere.17 The process of thermal decomposition of metal acetates, required in the production of catalysts, has been shown to be complex and quite sensitive to heat treatment conditions.18–20 Auto-reduction during heat treatment under inert atmospheres, combined with the potential of metal acetates to sublime,17 is considered to control the size of metal particles and their interaction with the support.
In this edge article, we show that highly active catalysts can be produced from chloride free precursors by the physical grinding of Au and Pd acetate salts with pre-formed supports. Bimetallic AuPd particles have been produced for the first time using this process. We use the catalysts prepared in this way to extend our earlier studies focusing on key reactions, namely the oxidation of benzyl alcohol and the direct synthesis of hydrogen peroxide. The removal of aqueous chloride ions from the preparation medium significantly decreases the waste material and ensures that the catalyst preparation is completed in a greener manner. Furthermore, we show that the removal of the chloride from the preparation step and therefore from the final catalyst, leads to a significant enhancement in performance as compared to previously reported catalysts. The origins of these effects are discussed.
Results and discussion
Palladium(II) acetate (Sigma Aldrich 99.9%) and gold(III) acetate (Alfa Aesar 99.9%) were added to the support (TiO2, Degussa, P25; SiO2, Matrix Silica 60 Fisher; carbon, G60, Aldrich) and the mixture was ground manually in a pestle and mortar for times varying between 1 and 20 min. The mass of acetate salts used was that required to give a 5 wt% total metal loading. For ease of comparison, the bimetallic catalysts were prepared with the metals in a 1:1 weight ratio and 5% metal by weight. The resultant mixture was placed in a 4 inch ceramic boat and heat treated at 350 °C for 2 h in a tubular furnace under flowing helium (ramp rate 1–20 °C). Details of catalytic testing for benzyl alcohol oxidation and hydrogen peroxide formation have been reported previously8 and are provided with catalyst characterisation methods in the ESI.†
All catalysts were heat treated under flowing helium at 350 °C, to facilitate the decomposition of the acetate ligands to form metal particles, as proposed by Lin et al.17 Heat treatments were not performed under an O2 containing atmosphere, as is conventional for standard catalyst preparations, due to the exceptionally exothermic decomposition of gold acetate.21
Thermal gravimetric analysis, performed under nitrogen, of the individual salts with and without the supports, and the combined salts with and without the supports was carried out (ESI Fig. S1–S4†). The decomposition temperatures of the gold and palladium salts corresponded well with those quoted in the literature,17,21 at 180 °C and 190 °C respectively. Gold acetate had a low temperature weight loss associated with adsorbed water. The close proximity of the two acetate decomposition temperatures could be a beneficial factor for the formation of alloyed AuPd materials. When the salts were combined (with a 1:1 wt ratio) there was no change in the decomposition kinetics or temperature of the two salts. The addition of either support material slowed the rate of acetate decomposition, implying an interaction exists between the supports and dispersed metal acetates, although reduced heat transfer rates, because of dilution by the support material could also be playing some role. Also noted was the splitting of the both metal acetate decomposition profiles into two distinct mass loss regions, in the presence of the TiO2 support. The lower temperature mass loss correlates with the bulk salt decomposition, while the higher temperature mass loss re-enforces the assertion of stable dispersed metal acetate salts. In addition, to the aforementioned effects, the TGA of palladium acetate with the carbon support showed mass loss occurring 20 °C lower than in the bulk salt alone. The exotherm associated with the acetate decomposition, noted in DTA analysis, also exhibited a similar temperature shift. The enhancement of the decomposition process, previously tentatively suggested,17 can be taken as a further indication of a complex interaction between the supports and dispersed metal acetates.
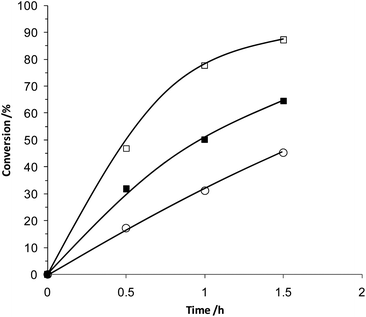
Initial catalysts were prepared using titania as we have shown it to be an extremely effective support for Au and Pd nanocrystals, capable of generating excellent catalytic materials for both oxidation and reduction reactions.9,11 We have previously reported that catalysts prepared by deposition precipitation (DP), which have low chloride content due to deliberate washing steps designed to remove Na and Cl, have a much higher initial activity for benzyl alcohol oxidation, when compared to catalysts prepared by impregnation (IMP).9 This is shown in the time-on-line data presented in Fig. 1. The Au + Pd catalyst prepared by physical grinding (PG) had a comparable initial activity to the DP catalyst.
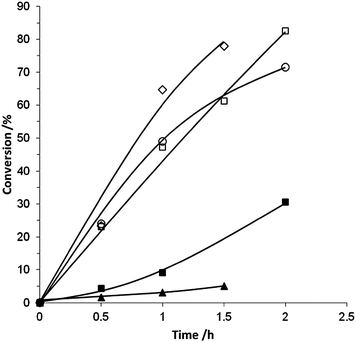 | ||
| Fig. 1 The conversion of benzyl alcohol by catalysts prepared using various preparation techniques: ▲ Au/TiO2 (PG), ■ AuPd/TiO2 (IMP), ○ AuPd/TiO2 (DP), □ AuPd/TiO2 (PG), ◇ Pd/TiO2 (PG). | ||
The TOFs (mol substrate converted per mol of metal per hour) determined for the different catalysts emphasise the high activity of the physically ground catalyst (Table 1). The catalysts prepared by DP show an initial TOF at 0.5 h of 20611 h−1 whereas the PG catalyst shows an initial TOF at 0.5 h of 27200 h−1. PG monometallic catalysts were also investigated with the 5 wt% Au/TiO2 material showing poor activity (TOF at 0.5 h of 2100 h−1) and the 5 wt% Pd/TiO2 displaying significant activity (TOF at 0.5 h of 15347 h−1). Although the conversion profiles (Fig. 1) of the pure Pd and the AuPd bimetallic catalysts are similar, when considered in terms of the initial TOF at 0.5 h, the bimetallic catalyst is the most active. This indicates a small synergistic effect occurring between the metals, which is consistent with results we have previously published on catalysts prepared by the IMP and DP methods.9–11 The bimetallic catalyst also displayed the best selectivity towards benzaldehyde when compared at similar conversion levels (Fig. 2), once again suggesting there is a beneficial interaction between the two metals.
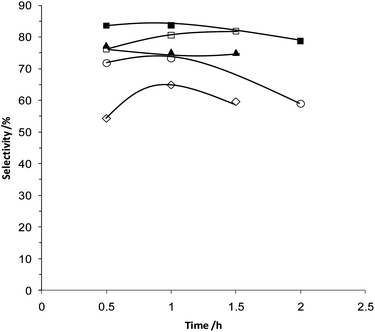 | ||
| Fig. 2 Selectivity towards benzaldehyde using catalysts prepared by various techniques: ▲ Au/TiO2 (PG), ■ AuPd/TiO2 (IMP), ○ AuPd/TiO2 (DP), □ AuPd/TiO2 (PG), ◇ Pd/TiO2 (PG). | ||
Catalysts that are active for the oxidation of benzyl alcohol have also previously been shown to be active for the direct synthesis of hydrogen peroxide.10,11 The activity of the 2.5% Au + 2.5% Pd/TiO2 catalyst, prepared by physical grinding, was 89 mol H2O2 kg−1 h−1 which compares favourably with previously reported data for catalysts prepared by impregnation with the same metal composition, which have an activity of 64 mol kg−1 h−1.11 The monometallic catalysts broadly follow the same trend as observed with the oxidation of benzyl alcohol. The Au catalyst was significantly less active than the analogous impregnation catalyst (7 mol kg−1 h−1 IMP (ref. 11) vs. 1 mol kg−1 h−1 PG) suggesting that the gold is not being dispersed to any great extent in the physically ground catalyst. However, it should be noted that characterisation studies of IMP catalysts have previously shown a considerable number of relatively large (35–80 nm) metal particles.11 This implies that the particle size distributions from the two preparation methods are substantially different, with the IMP catalyst having a broader distribution and a greater number of small particles. Confirmation of this hypothesis required characterisation using techniques that provide a measure of average crystallite size, such as XRD analysis as discussed below. The reverse trend in activity was observed for the 5% Pd/TiO2 catalysts, with the physically ground catalyst showing greater activity than the impregnation catalyst (30 mol kg−1 h−1 IMP (ref. 11) vs. 45 mol kg−1 h−1 PG), which suggests that an improved distribution of the Pd is being achieved by the PG preparation method, as has been noted previously.17 It has been reported that the incorporation of only a small amount of Au into a Pd-rich particle leads to a significant increase in the activity of the catalyst.22 The measured increase in activity from 45 to 89 mol kg−1 h−1 suggests that some Au may be getting incorporated into the Pd species in our physically ground catalysts.
X-Ray diffraction of the mono- and bimetallic samples produced by PG, along with data from a comparable standard IMP bimetallic catalyst, are shown in Fig. 3 with calculated parameters shown in Table 2. Refinement of the monometallic XRD patterns showed metallic Au and Pd and was in good agreement with interpretations of the crystallite size derived from catalytic testing. The average Au crystallite size was significantly larger than that seen in the comparable IMP catalyst. This illustrates the overall poorer dispersion of Au metal over the TiO2 given by the PG method. The small mean crystallite size (7 nm) of the Pd particles produced by PG indicated a far greater intrinsic ability of this metal to disperse over the support. The bimetallic AuPd catalyst produced by acetate decomposition showed reflections associated with two crystalline cubic phases, with unit cell volumes closely associated with Au and Pd. Both phases have an observable shift in unit cell volumes compared to the respective comparable monometallic systems. However, no reflections associated with the unit cell size of a bulk 1:1 AuPd alloy (63.04 Å) were observed.23 This suggests some limited alloying, with a small incorporation of one element into the bulk of the other or by the formation of a core shell structure as seen previously for IMP AuPd/TiO2 catalysts.11,19 Though no reflections indicative of a homogenous AuPd alloy were observed, a proportion of nanoparticles that are below the detectability limit of the XRD technique could still be present. The calculated weight contribution of the Pd and Au phases in the PG bimetallic catalyst accounts for only 60% of the metal loading (Table 2), while energy dispersive X-ray analysis obtained from an extended area of the sample (see ESI, Fig S5†) suggested that the catalyst had the anticipated nominal metal weight loading (4.9 wt% ± 0.2%), implying the presence of additional sub-5 nm particles that were not being detected by XRD.
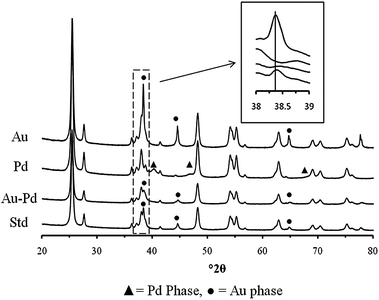 | ||
| Fig. 3 X-Ray diffraction of mono- and bimetallic samples supported on TiO2 produced by PG. The comparative standard (Std) is a bimetallic AuPd catalyst prepared by impregnation (IMP). | ||
| Sample | Metal phase | Unit cell volume/Å3 | Average crystallite size/nm | Weight fraction of phase (%) |
|---|---|---|---|---|
| a Errors: unit cell volume ±0.1 Å3, crystallite size ±2 nm, weight fraction ±0.5%. | ||||
| Au/TiO2 (PG) | Au | 67.79 | 57 | 4.4 |
| Pd/TiO2 (PG) | Pd | 59.03 | 7 | 4.7 |
| Au-Pd/TiO2 (PG) | Pd | 59.65 | 4 | 1.2 |
| Au | 67.26 | 62 | 2.0 | |
| Au-Pd/TiO2 (Std-IMP) | Pd | n/a | n/a | n/a |
| Au | 67.70 | 34 | 2.2 | |
Scanning transmission electron microscopy (STEM) analysis of the PG 2.5% Au + 2.5% Pd/TiO2 catalyst (Fig. 4) confirms the deductions from XRD analysis. Large, gold rich, particles of ca. 50 nm were observed. In addition, the technique provided confirmation of the presence of a significant population of 2–5 nm particles that are not detectable by XRD analysis. From XEDS analysis these particles have significant gold and palladium contributions, demonstrating alloying. These sub-5 nm AuPd particles, which were previously alluded to by the missing weight contributions in XRD analysis, are strongly suspected to be responsible for the high catalytic activity, as previously reported in the literature.9
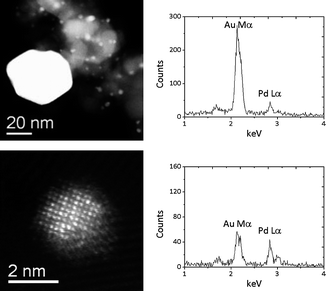 | ||
| Fig. 4 Representative STEM HAADF images and corresponding XEDS analysis of particles from 2.5 wt% Au + 2.5 wt% Pd/TiO2 catalyst prepared by the physical mixing of acetate salts. The top row shows data from a typical large Au-rich particle (∼50 nm in size), whereas the lower row shows data from a 2 nm AuPd nm alloy particle. | ||
Fig. 5 shows the Pd(3d) spectral region for a PG 2.5% Au + 2.5% Pd/TiO2 catalyst before and after heat treatment. For the unheated catalysts we observe a Pd(3d5/2) peak at 336.9 eV, which shifts to 336.0 eV after heat treatment, the Pd(3d5/2) binding energy for metallic Pd is ca. 335.0 eV. The 336 eV peak is close to that expected for PdO generated by the decomposition of the acetate, but the presence of surface PdC must be considered, as observed after reactions of Pd foil with several small organic molecules.24 A core–shell effect with carbidic species has been observed previously with metals produced from the decomposition of metal acetates.20 The comparable selectivity to benzaldehyde of the PG, DP and IMP derived catalysts, shown in Fig. 2, suggests that the carbidic species did not have an obvious detrimental effect on selectivity. As a result of the changes in the C(1s) profile from the different treatments, we have referenced all binding energies to the Ti(2p3/2) peak from the support, taken to be 458.2 eV.25 The binding energy difference between Pd2+ in the acetate and Pd2+ in PdO arises from a combination of different degrees of charge transfer between the Pd and ligand, and differences in final state relaxation effects. Quantification of the integrated peak intensities (Table 3) shows that there is an excess of surface Pd over surface Au, especially after heating the dried catalyst in He. This shows that the Pd disperses far more successfully than Au on heat treatment. Alternatively the high Pd surface contribution may indicate some development of a some core–shell morphology,11,19 although this was not observed in the corresponding STEM-HAADF imaging analysis.
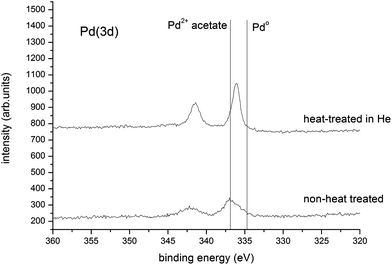 | ||
| Fig. 5 Pd(3d) spectra for the physically ground acetate salts with TiO2 and the catalyst after heating in He. | ||
To further extend the initial work, a range of bimetallic catalysts were also prepared using a variety of other supports that had previously been shown to give active catalysts for these reactions. A comparison of catalysts prepared on different supports for the oxidation of benzyl alcohol is shown in Fig. 6. It is clear that the physical grinding method can produce catalysts that are active for benzyl alcohol oxidation on all the support materials tested. These catalysts are more active than the previously reported materials prepared by impregnation.10 The stability of the carbon supported catalyst, as it was the most active, was tested by re-using it for the oxidation of benzyl alcohol (ESI Table 1†). These catalysts proved to be re-usable within experimental error. The equivalent monometallic catalysts on C were also prepared and tested for benzyl alcohol oxidation (Fig. 7).
 | ||
| Fig. 6 Comparison of 2.5% Au + 2.5% Pd PG catalysts on different support materials for the oxidation of benzyl alcohol: ■ TiO2, ○ SiO2, □ G60. | ||
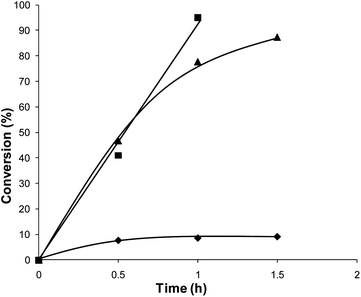 | ||
| Fig. 7 Conversion of benzyl alcohol with mono- and bimetallic PG catalysts supported on carbon: ◆ Au, ▲ AuPd, ■ Pd. | ||
In common with the TiO2 supported catalysts described earlier, the Pd and AuPd catalysts had significantly higher initial activity than the Au catalyst, with the bimetallic catalyst showing the highest activity (Table 4). The effect was observed to be greater than that for the equivalent TiO2 supported catalysts prepared by PG.
| Metal (5% total) | TOF (calculated at 0.5 h) |
|---|---|
| Au | 9340 |
| Pd | 26900 |
| AuPd | 39900 |
The trend found for H2O2 synthesis (Table 5) is that for all the supports tested, the activity obtained is similar to those reported previously for catalysts prepared by impregnation methods.11,26,27 However, in most cases the productivity was found to be greater with the physical grinding method, with the exception of the carbon supported catalyst. In the latter case, the lower productivity can be explained by the significantly higher hydrogenation activity when compared to the comparable material prepared by impregnation. Re-use tests were carried out for all these catalysts and the productivity for the TiO2 and the carbon supported catalyst displayed no decrease in activity upon the second use (ESI Table 1†). The comparable silica supported catalyst, however, did show a decrease in productivity on the second use, indicating that the metal particles are not stable on this particular support.
| Support | Impregnation | Physical grinding |
|---|---|---|
| Productivity (mol H2O2 per kg-cat per h) | ||
| C (G60) | 110 | 102 |
| TiO2 | 64 | 90 |
| SiO2 | 108* | 142* |
| Hydrogenation activity (mol H2O2 per kg-cat per h) | ||
|---|---|---|
| C (G60) | 5% | 672 (33%) |
AN XRD analysis of samples produced by acetate decomposition on the G60 carbon support is shown in Fig. 8. In common with the Au/TiO2 sample, the average crystallite size of the monometallic Au/C is relatively large (ca. 30 nm), as compared to that produced by other catalyst preparation techniques. The monometallic Pd supported on carbon showed two distinct phases with different unit cell sizes. The conventional f.c.c. Pd phase had a unit cell size of 59.40 Å3 while a second phase had an expanded unit cell of 63.09 Å3. The expanded unit cell is indicative of incorporation of additional species into the Pd unit cell, such as interstitial carbon.28–30 As expected there was no evidence of interstitial C incorporation into the f.c.c. Au phase, since these two components are known to be immiscible.31 The 2.5% Au + 2.5% Pd/C catalysts produced by acetate decomposition once again resulted in the formation of distinct Au and Pd crystals, as noted for the TiO2 supported materials. Unfortunately, the possible presence of the expanded Pd phase in the monometallic sample makes unambiguous identification of an AuPd alloy difficult to confirm by XRD. However, the synergistic effect noted for benzyl alcohol oxidation and the direct H2O2 reaction found for the PG carbon supported bimetallic catalyst indicates that a degree of alloying has indeed occurred.
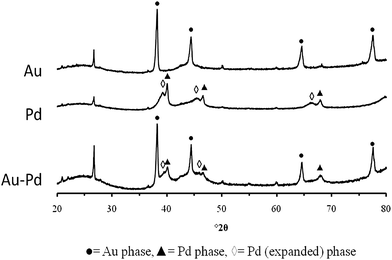 | ||
| Fig. 8 XRD analysis of mono- and bimetallic samples produced by acetate decomposition on a carbon support. | ||
Scanning transmission electron microscopy of the carbon supported AuPd catalysts demonstrated the presence of sub-5 nm particles that are not detectable by XRD. The 2.5% Au+2.5% Pd/C catalyst (Fig. 9) was found to comprise of large (ca.0.2 μm) Au-rich and some large (ca.0.1 μm) Pd-rich particles. However smaller sub-5 nm AuPd random alloy particles of widely varying composition and size were also frequently observed (Fig. 10). The existence of such alloyed nanoparticles would explain the synergistic effect noted from the catalytic data. STEM analyses of the monometallic Au/C and Pd/C samples are shown in ESI Fig. S6 and S7† respectively and broadly corroborate the XRD findings for the presence of carbidic Pd species. This supports the XPS in showing the presence of PdC species. However, the observation of PdC by XRD and STEM shows that it is not exclusively a surface species.
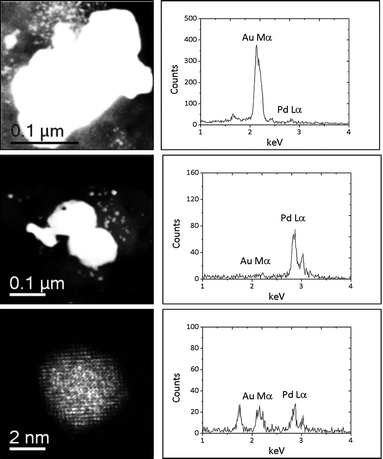 | ||
| Fig. 9 Representative HAADF images and corresponding XEDS spectra from the 2.5 wt% Au + 2.5 wt% Pd/C catalyst produced by physical grinding of acetate salts. The top row shows a large Au particle (∼250 nm in size), whereas the middle row shows some large (∼100 nm) Pd particles. In addition, AuPd random alloy nanoparticles are also present as demonstrated by the 4 nm particle shown in the bottom row. | ||
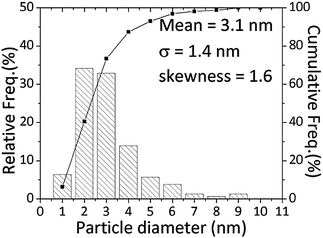 | ||
| Fig. 10 Size distribution of sub-10 nm AuPd alloy particles in the 2.5 wt% Au + 2.5 wt% Pd/C catalyst produced by physical grinding. Note: many much larger Au-rich and Pd-rich particles were also observed but are not included in this histogram. | ||
Physical grinding of the metal acetate precursor with the support provides improved macroscale distribution of the precursors on the support, prior to the sublimation stage which is induced by the heat treatment step of the synthesis. This then leads to the observed mixture of pure-Au, pure-Pd and AuPd alloy nanoparticles. In order to ascertain the importance of the initial mixing of the support and the metal acetate precursors, a series of AuPd/C catalysts were prepared with increasing grinding time. These catalysts were subsequently heat treated and tested for benzyl alcohol oxidation and hydrogen peroxide formation as shown in Tables 6 and 7 respectively. It is clear that the conversion of the benzyl alcohol (Table 6) is similar for all grinding times with the possible exception of the 1 min sample. Hydrogen peroxide synthesis tests (Table 7) showed the same productivity for all the grinding times attempted. This indicates the most important redistribution of the metal occurs during the heat treatment stage rather than the grinding stage, once a certain level of basic mixing has been achieved.
| Grind time/min | Conversion (%) | Benzaldehyde selectivity (%) | ||
|---|---|---|---|---|
| 0.5 h | 1 h | 0.5 h | 1 h | |
| 1 | 40.5 | 69.5 | 77.9 | 79.3 |
| 5 | 47.5 | 77.0 | 77.8 | 79.1 |
| 10 | 50.8 | 74.3 | 82.4 | 79.0 |
| 20 | 47.7 | 78.3 | 83.6 | 85.3 |
| Grind time/min | H2O2 productivity/mol kg−1 h−1 |
|---|---|
| 1 | 100 |
| 5 | 102 |
| 10 | 102 |
| 20 | 104 |
Conclusions
We have shown the first example of the successful use of alloy catalysts prepared by heat treatment of physically mixed metal acetate precursors. Supported gold, palladium and gold–palladium catalysts have been prepared and shown to be active for two key reactions known to be catalysed by these metal systems. This simple, reproducible and scalable preparation method ensures the catalysts prepared are chloride free. The study provides a base for future work into the effect of chloride on benzyl alcohol oxidation and hydrogen peroxide formation. Catalytic data supported by XRD and STEM characterisation indicate that some degree of alloying between two metal components can be achieved using this preparation method, which has not been reported previously. We have also demonstrated that these catalysts are more active than equivalent catalysts prepared by the impregnation route for key redox processes.Notes and references
- A. K. Sinha, S. Seelan, S. Tsubota and M. Haruta, Angew. Chem., Int. Ed., 2004, 43, 1546 CrossRef CAS.
- M. D. Hughes, Y.-J. Xu, P. Jenkins, P. McMorn, P. Landon, D. I. Enache, A. F. Carley, G. A. Attard, G. J. Hutchings, F. King, E. H. Stitt, P. Johnston, K. Griffin and C. J. Kiely, Nature, 2005, 437, 1132 CrossRef CAS.
- L. Kesavan, R. Tiruvalam, M. H. A. Rahim, M. I. bin Saiman, D. I. Enache, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, S. H. Taylor, D. W. Knight, C. J. Kiely and G. J. Hutchings, Science, 2011, 311, 195 CrossRef.
- S. Biella, L. Prati and M. Rossi, J. Catal., 2002, 206, 242 CrossRef CAS.
- F. Porta and L. Prati, J. Catal., 2004, 224, 397 CrossRef CAS.
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 405 CrossRef CAS.
- M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301 CrossRef CAS.
- P. Landon, P. J. Collier, A. J. Papworth, C. J. Kiely and G. J. Hutchings, Chem. Commun., 2002, 2058 RSC.
- P. J. Miedziak, Q. He, J. K. Edwards, S. H. Taylor, D. W. Knight, B. Tarbit, C. J. Kiely and G. J. Hutchings, Catal. Today, 2011, 163, 47 CrossRef CAS.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362 CrossRef CAS.
- J. K. Edwards, B. E. Solsona, P. Landon, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, J. Catal., 2005, 236, 69 CrossRef CAS.
- A. Schulz and M. Hargittai, Chem.–Eur. J., 2001, 7, 3657 CrossRef CAS.
- H. S. Oh, J. H. Jang, C. K. Costello, W. M. Wang, S. R. Bare, H. H. Kung and M. C. Kung, J. Catal., 2002, 210, 375 CrossRef CAS.
- I. Dobrosz-Gómez, I. Kocemba and J. M. Rynkowski, Appl. Catal., B, 2009, 88, 83 CrossRef.
- H. H. Kung, M. C. Kung and C. K. Costello, J. Catal., 2003, 216, 425 CrossRef CAS.
- F. Moreau, G. C. Bond and A. O. Taylor, J. Catal., 2005, 231, 105 CrossRef CAS.
- Y. Lin, K. A. Watson, M. J. Fallbach, S. Ghose, J. G. Smith, D. M. Delozier, W. Cao, R. E. Crooks and J. W. Connell, ACS Nano, 2009, 3, 871 CrossRef CAS.
- S. A. Kondrat, T. E. Davies, Z. Zu, P. Boldrin, J. K. Bartley, A. F. Carley, S. H. Taylor, M. J. Rosseinsky and G. J. Hutchings, J. Catal., 2011, 281, 279 CrossRef CAS.
- J. C. De Jesus, I. González, A. Quevedo and T. Puerta, J. Mol. Catal. A: Chem., 2005, 228, 283 CrossRef CAS.
- S. V. Pol, V. G. Pol, I. Felner and A. Gedanken, Eur. J. Inorg. Chem., 2007, 2089 CrossRef CAS.
- S. D. Bakrania, G. K. Rathore and M. S. Wooldrigde, J. Therm. Anal. Calorim., 2009, 95, 117 CrossRef CAS.
- J. K. Edwards, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Faraday Discuss., 2008, 138, 225 RSC.
- A. Maeland and T. B. Flanagan, Can. J. Phys., 1964, 42, 2364 CrossRef CAS.
- D. Teschner, J. Borsodi, A. Wootsch, Z. Révay, M. Hävecker, A. Knop-Gericke, S. D. Jackson and R. Schlögl, Science, 2008, 320, 86 CrossRef CAS.
- www.cf.ac.uk/chemy/staffinfo/xpsaccess/database/tio2p25.html .
- J. K. Edwards, A. Thomas, B. E. Solsona, P. Landon, A. F. Carley and G. J. Hutchings, Catal. Today, 2007, 122, 397 CrossRef CAS.
- J. K. Edwards, B. Solsona, E. N. Ntainjua, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037 CrossRef CAS.
- B. Ziemecki, G. A. Jones, D. G. Swartzfager, R. L. Harlow and J. Faber, J. Am. Chem. Soc., 1985, 107, 4547 CrossRef.
- M. Maciejewski and A. Baiker, Pure Appl. Chem., 1995, 67, 1879 CrossRef CAS.
- M. Beck, M. Ellner and E. J. Mittemeijer, Acta Mater., 2001, 49, 985 CrossRef CAS.
- H. Okamoto and T. B. Massalski, Bull. Alloy Phase Diagrams, 1984, 5, 378 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and supplementary figures. See DOI: 10.1039/c2sc20450a |
| This journal is © The Royal Society of Chemistry 2012 |