A mononuclear carboxylate-rich oxoiron(IV) complex: a structural and functional mimic of TauD intermediate ‘J’†
Aidan R.
McDonald
a,
Yisong
Guo
b,
Van V.
Vu
a,
Emile L.
Bominaar
*b,
Eckard
Münck
*b and
Lawrence
Que
Jr.
*a
aDepartment of Chemistry and Center for Metals in Biocatalysis, University of Minnesota, 207 Pleasant Street S.E., Minneapolis, MN 55455, U. S. A.
bDepartment of Chemistry, Carnegie Mellon University, Mellon Institute, 4400 Fifth Ave., Pittsburgh, PA 15213, U. S. A.
First published on 20th February 2012
Abstract
The pentadentate ligand nBu-P2DA (2(b), nBu-P2DA = N-(1′,1′-bis(2-pyridyl)pentyl)iminodiacetate) was designed to bind an iron center in a carboxylate-rich environment similar to that found in the active sites of taurine dioxygenase (TauD) and other α-ketoglutarate-dependent mononuclear non-heme iron enzymes. The iron(II) complex nBu4N[FeII(Cl)(nBu-P2DA)] (3(b)-Cl) was synthesized and crystallographically characterized to have a 2-pyridine-2-carboxylate donor set in the plane perpendicular to the Fe–Cl bond. Reaction of 3(b)-Cl with N-heterocyclic amines such as pyridine or imidazole yielded the N-heterocyclic amine adducts [FeII(N)(nBu-P2DA)]. These adducts in turn reacted with oxo-transfer reagents at −95 °C to afford a short-lived oxoiron(IV) complex [FeIV(O)(nBu-P2DA)] (5(b)) in yields as high as 90% depending on the heterocycle used. Complex 5(b) exhibits near-IR absorption features (λmax = 770 nm) and Mössbauer parameters (δ = 0.04 mm s−1; ΔEQ = 1.13 mm s−1; D = 27 ± 2 cm−1), characteristic of an S = 1 oxoiron(IV) species. Direct evidence for an Fe![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of 1.66 Å was found from EXAFS analysis. DFT calculations on 5(b) in its S = 1 spin state afforded a geometry-optimized structure consistent with the EXAFS data. They further demonstrated that the replacement of two pyridine donors in [FeIV(O)(N4Py)]2+ (N4Py = N,N-(bis(2-pyridyl)methyl)N-bis(2-pyridylmethyl)amine) with carboxylate donors in 5(b) decreased the energy gap between the ground S = 1 and the excited S = 2 states, reflecting the weaker equatorial ligand field of 5(b) and accounting for its larger D value. Complex 5(b) reacted readily with dihydrotoluene, methyldiphenylphosphine and ferrocene at −60 °C, and in all cases was approximately a 5-fold more reactive oxidant than [FeIV(O)(N4Py)]2+. The reactivity differences between these two complexes may arise from a combination of electronic and steric factors. Carboxylate-rich 5(b) represents the closest structural mimic reported thus far of the oxoiron(IV) intermediate (‘J’) found in TauD and provides us with vital insights into the role carboxylate ligands play in modulating the spectroscopic and reactivity properties of the non-heme oxoiron(IV) moiety.
O bond of 1.66 Å was found from EXAFS analysis. DFT calculations on 5(b) in its S = 1 spin state afforded a geometry-optimized structure consistent with the EXAFS data. They further demonstrated that the replacement of two pyridine donors in [FeIV(O)(N4Py)]2+ (N4Py = N,N-(bis(2-pyridyl)methyl)N-bis(2-pyridylmethyl)amine) with carboxylate donors in 5(b) decreased the energy gap between the ground S = 1 and the excited S = 2 states, reflecting the weaker equatorial ligand field of 5(b) and accounting for its larger D value. Complex 5(b) reacted readily with dihydrotoluene, methyldiphenylphosphine and ferrocene at −60 °C, and in all cases was approximately a 5-fold more reactive oxidant than [FeIV(O)(N4Py)]2+. The reactivity differences between these two complexes may arise from a combination of electronic and steric factors. Carboxylate-rich 5(b) represents the closest structural mimic reported thus far of the oxoiron(IV) intermediate (‘J’) found in TauD and provides us with vital insights into the role carboxylate ligands play in modulating the spectroscopic and reactivity properties of the non-heme oxoiron(IV) moiety.
Introduction
Carboxylate ligation plays an important role in modulating the structural and electronic properties of metal sites found in an array of oxygen activating metalloenzymes. In particular non-heme iron enzymes, which catalyze a wide variety of oxidative transformations, rely upon relatively weak field carboxylate donors to tune the reactivity of their iron active sites.1–3 Many structural investigations have demonstrated the 2-His-1-carboxylate facial triad to be the common structural motif for the active sites of these enzymes.4–6 Members of this superfamily catalyze, amongst others, protein modification, alkylated DNA/RNA repair, antibiotic biosynthesis, and oxidative degradation of aromatic compounds.3,5 The range of oxidative transformations involved in these processes is equally broad: hydroxylation and halogenation of aliphatic C–H bonds, desaturation of alkanes, cis-dihydroxylation of arenes, C–C bond cleavage, and olefin epoxidation. Given the tremendous scope of carboxylate-ligated non-heme iron enzymes, it is surprising how little we understand of the role of the carboxylate donor in these enzymes.Enzymes that utilize the 2-His-1-carboxylate facial triad, such as the α-ketoglutarate (α-KG) dependent dioxygenases, pterin dependent hydroxylases, and isopenicillin N synthase (IPNS), cycle through a common catalytic mechanism that involves formation of a substrate oxidizing oxoiron(IV) species.7–22 The oxoiron(IV) moiety, always found in the S = 2 spin state, is of particular interest because it is an oxidant capable of oxidizing very strong C–H bonds. In the case of the α-KG-dependent enzyme taurine dioxygenase (TauD), the oxoiron(IV) moiety is postulated to be ligated by two carboxylate ligands cis to the oxo ligand (Scheme 1),21 while for phenylalanine hydroxylase (PheH, a pterin-dependent hydroxylase) the oxoiron(IV) species is believed to have a single bidentate carboxylate ligand cis to the oxo (Scheme 1).22 On the other hand, the high-valent intermediate of IPNS is hypothesized to contain a thiolate group ligated cis to the oxo ligand with a carboxylate ligand bound trans to the oxo unit (Scheme 1).18,20 Thus these oxoiron(IV) oxidants all contain at least two weak-field donors cis to the oxo group; they can play a significant role in the modulation of the spin state and reactivity of the metal–oxo unit by altering the energies and thus occupancies of the dxy and dx2−y2 orbitals associated with the metal center. In addition to tuning the electronic properties of the oxoiron(IV) moiety, the carboxylate ligand can act as a hydrogen-bond acceptor for an iron-ligated H2O molecule as found in CytC3 halogenase.23 An equally important aspect of anionic carboxylate ligands is that they neutralize the charge on the iron center.
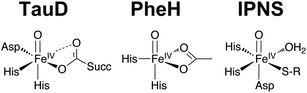 | ||
| Scheme 1 Putative oxoiron(IV) intermediates from members of the 2-His-1-carboxylate facial triad superfamily of mononuclear non-heme iron enzymes. | ||
We and others have used neutral nitrogen-rich polydentate ligands to generate a family of synthetic mononuclear non-porphyrin oxoiron(IV) complexes that act as functional mimics of the intermediates depicted in Scheme 1.24–38 Ligands that enforce an octahedral (oct) ligand field such as N,N-bis(2-pyridylmethyl)-N-bis(2-pyridyl)methylamine (N4Py, Scheme 2),30 tris(2-pyridylmethyl)amine (TPA, Scheme 2),29 and 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclodecane (TMC, Scheme 2)28 yielded intermediate spin (S = 1) oxoiron(IV) complexes whereas ligands that enforce a trigonal bipyramidal (tbp) ligand field such as 1,1,1-tris(2-[N2-(1,1,3,3-tetramethylguanidinium)]ethyl)amine (TMG3tren, Scheme 2)34,35 and 1,1,1-tris(2-(tert-butylurealato)ethyl)amine (H3buea, Scheme 2)36 yielded high spin (S = 2) oxoiron(IV) complexes. Examination of the crystal-field splitting diagrams for oct and tbp ligand fields shows that the energy difference between the dxy and dx2−y2 orbitals, and thus the equatorial ligand field, controls the spin state of a d4 metal–oxo complex. The equatorially nitrogen-rich oct oxoiron(IV) complexes apparently have a large dxy/dx2−y2 gap and thus spin pairing occurs yielding S = 1 complexes. The tbp geometry naturally enforces a weak equatorial ligand field resulting in degenerate, or nearly degenerate, dxy and dx2−y2 orbitals, yielding S = 2 complexes. The precise ligand field geometry in the enzymatic oxoiron(IV) intermediates depicted in Scheme 1 is not known; however the iron centers of all intermediates trapped thus far have been S = 2.18 Computational studies based on the spectroscopic properties of the isolated oxoiron(IV) intermediate ‘J’ from TauD suggest both oct or tbp geometries are plausible.21 Nonetheless, it is reasonable to assume that the equatorial weak-field carboxylate donors in enzymatic oxoiron(IV) intermediates ensure that the iron center is S = 2. This is an important point as DFT calculations suggest that the S = 2 oxoiron(IV) moiety can be a considerably faster oxidant than the S = 1.39–46 It should also be noted that the above mentioned nitrogen-rich synthetic oxoiron(IV) compounds are all charged complexes, in contrast to the enzymatic carboxylate-rich oxoiron(IV) intermediates which are, in general, neutral species. The reactivity of a metal–oxo species towards hydrogen atom transfer (HAT) can be dependent on the redox potential of the metal–oxo moiety and on the pKa of the metal-hydroxo product.47,48 Given that anionic donors will lower the redox potential of an oxoiron(IV) moiety while potentially yielding a less acidic hydroxoiron(III) product, when compared to charged nitrogen-rich oxoiron(IV) species, it is important to gauge the role anionic donors play in modulating the reactivity of oxoiron(IV) species.
 | ||
| Scheme 2 Structures of some synthetic oxoiron(IV) complexes. | ||
To date, no synthetic oxoiron(IV) complex containing more than one anionic carboxylate donor has been reported; therefore no genuine structural mimics of the oxoiron(IV) intermediates depicted in Scheme 1 are available. To address this challenge, suitable iron(II) precursors need to be identified. While there are a number of well characterized monoiron(II) complexes with two carboxylate donors reported in the literature,49–57 none of these has been demonstrated to serve as a precursor for an oxoiron(IV) complex. Towards this end, we have designed and synthesized the pentadentate ligand N-(1′,1′-bis-(2-pyridyl)methyl)iminodiacetate (P2DA, Scheme 3) and characterized corresponding mononuclear iron(II) complexes. We also describe the conversion of one of these iron(II) complexes to its oxoiron(IV) derivative and report a detailed spectroscopic characterization of the latter. This oxoiron(IV) complex represents the first structural mimic for the iron center of intermediate ‘J’ of TauD and provides insights into how carboxylate ligands can modulate the reactivity of the oxoiron(IV) unit.
 | ||
| Scheme 3 Synthesis of P2DA ligands 2(a) and 2(b). | ||
Results and discussion
Ligand and complex synthesis
N-(1′,1′-bis-(2-pyridyl)methyl)iminodiacetate (P2DA) ligands 2(a) and 2(b), were synthesized using standard synthetic protocols for the introduction of carboxymethyl functionalities onto amines 1(a) and 1(b) (Scheme 3).58 The synthesis of 1(b) was necessitated by the need to generate an iron(II) complex that was soluble in common organic solvents, and was accomplished by introducing an n-butyl functionality onto 1(a). This was achieved by deprotonation of the methine group of 1(a) followed by coupling of the formed carbanion with 1-bromobutane to yield 1(b) quantitatively (see Scheme 3).59 Compounds 1(a) and 1(b) were reacted with 3 equiv. of ethyl iodoacetate in the presence of 4 equiv. of K2CO3 to yield diethyl-N-(1′,1′-bis-(2-pyridyl)alkyl)iminodiacetate diesters. Hydrolysis of the diester compounds with KOH in H2O/tetrahydrofuran (THF) yielded the desired dipotassium salts in near quantitative yields. Compound 2(a) (K2P2DA) was obtained in 55% overall yield, whereas poorer overall yield (15%) was obtained for compound 2(b) (K2nBu-P2DA) due to significant losses during column chromatography of the diester intermediate.Combining ligand 2(a) with FeCl2 in methanol (CH3OH) under anaerobic conditions at room temperature resulted in the rapid precipitation (10 min) of a red crystalline material (3(a), Scheme 4A). The same observation was made when the reaction was carried out in dimethylformamide (DMF). Furthermore, alternative iron(II) sources such as [Fe(OTf)2(NCCH3)2] (OTf = trifluoromethanesulfonate) or [Fe(NCCH3)4](BF4)2 also yielded the same insoluble material. Complex 3(a) was insoluble in common organic solvents (such as CH3OH, CH2Cl2, acetonitrile (CH3CN), or DMF), but did dissolve in H2O yielding a pale yellow solution. Electrospray ionization mass spectrometry (ESI-MS) of 3(a) showed the expected mass peak at m/z = 394.2 corresponding to the (K[FeII(P2DA)])+ ion (Figure S1, Supporting Information†). 1H NMR analysis (D2O, Fig. 1, left) showed paramagnetically shifted resonances, comparable to those observed in the S = 2 iron(II) complexes [FeII(Cl)(N4Py)]+, [FeII(TPA)(Cl)2], and [FeII(6-Me3-TPA)(NCCH3)2]2+ (6-Me3-TPA = tris(6-methyl-2-pyridylmethyl)amine).60–62 The observation of seven resonances would suggest that 3(a) in aqueous solution is a mononuclear iron(II) complex with a ligand-to-iron(II) ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. If multinuclear, carboxylate-bridged iron(II) complexes or non-integer ligand-to-metal ratios were present, more than seven resonances would be anticipated. FT-IR analysis of 3(a) in the solid state demonstrated very broad signals at ∼1620 and ∼1370 cm−1 assigned to the symmetric and asymmetric vibrations of the carboxylate moieties (Figure S2, Supporting Information†). None of the spectroscopic techniques we used allowed identification of the possible sixth ligand X depicted for 3(a) in Scheme 4. However, based on the elemental analysis data, the only reasonable formulation for 3(a) is K[FeII(OH)(P2DA)], so X is assigned to be hydroxide. Given its insolubility in organic solvents, 3(a) seemed unlikely to be an appropriate precursor from which to trap a reactive oxoiron(IV) species at lower temperatures. Our synthetic efforts thus shifted to a butylated P2DA derivative that afforded an iron(II) complex that was soluble in a number of organic solvents.
:1. If multinuclear, carboxylate-bridged iron(II) complexes or non-integer ligand-to-metal ratios were present, more than seven resonances would be anticipated. FT-IR analysis of 3(a) in the solid state demonstrated very broad signals at ∼1620 and ∼1370 cm−1 assigned to the symmetric and asymmetric vibrations of the carboxylate moieties (Figure S2, Supporting Information†). None of the spectroscopic techniques we used allowed identification of the possible sixth ligand X depicted for 3(a) in Scheme 4. However, based on the elemental analysis data, the only reasonable formulation for 3(a) is K[FeII(OH)(P2DA)], so X is assigned to be hydroxide. Given its insolubility in organic solvents, 3(a) seemed unlikely to be an appropriate precursor from which to trap a reactive oxoiron(IV) species at lower temperatures. Our synthetic efforts thus shifted to a butylated P2DA derivative that afforded an iron(II) complex that was soluble in a number of organic solvents.
![Synthesis of [FeII(P2DA)] complexes 3(a) and 3(b).](/image/article/2012/SC/c2sc01044e/c2sc01044e-s4.gif) | ||
| Scheme 4 Synthesis of [FeII(P2DA)] complexes 3(a) and 3(b). | ||
![1H NMR spectra of 3(a), [FeII(P2DA)] (left), 3(b)-Cl, K[FeII(Cl)(nBu-P2DA)] (right, the resonances at ∼1 and ∼26 ppm have two features) measured at 23 °C.](/image/article/2012/SC/c2sc01044e/c2sc01044e-f1.gif) | ||
| Fig. 1 1H NMR spectra of 3(a), [FeII(P2DA)] (left), 3(b)-Cl, K[FeII(Cl)(nBu-P2DA)] (right, the resonances at ∼1 and ∼26 ppm have two features) measured at 23 °C. | ||
The reaction between ligand 2(b) and FeCl2 in CH3OH afforded K[FeII(Cl)(nBu-P2DA)], which was converted by metathesis to the corresponding tetrabutylammonium salt 3(b)-Cl (60% yield) (Scheme 4(B)). No precipitation was observed, likely because the n-butyl tail enhanced the solubility of the formed iron(II) complex in CH3OH. ESI-MS analysis of compound 3(b)-Cl in the negative mode showed a mass peak corresponding to the [FeII(Cl)(nBu-P2DA)]− ion (m/z = 446.5) (Figure S3, Supporting Information†). 1H NMR analysis (CD2Cl2) of the K+ salt of 3(b)-Cl (Fig. 1, right, K+ salt used as the tetrabutylammonium counterion masked regions of the 1H NMR spectrum) showed ten paramagnetically shifted resonances characteristic of an S = 2 iron(II) complex similar to those observed for 3(a) (Fig. 1), consistent with the formulation of 3(b)-Cl as a mononuclear iron(II) complex with a ligand-to-iron(II) ratio of 1:1 for the same reasons as detailed above. The spectra obtained for complexes 3(a) and 3(b)-Cl showed minimal differences: a broad resonance at 140–150 ppm and four/five resonances between 20 and 70 ppm were observed. These similarities suggest to us that the H2O-soluble complex 3(a) has the same structural make-up as the organic soluble complex 3(b)-Cl but differs in the nature of the apical (sixth) ligand.60–62 FT-IR analysis of 3(b)-Cl demonstrated resonances at ν = 1631 and 1380 cm−1 assigned to the asymmetric and symmetric carboxylate vibrations (Figure S4, Supporting Information†), the energies of which match well with other end-on metal–carboxylate complexes.63
Crystals suitable for X-ray diffraction analysis were obtained from CH2Cl2/diethyl ether for the Ph4P+ salt of 3(b)-Cl. The structural analysis demonstrated the formation of a mononuclear iron(II) compound, with the pentadentate nBu-P2DA ligand binding the iron(II) center in a distorted octahedral fashion (Fig. 2). As anticipated, the carboxylate donors are monodentate and occupy the equatorial plane. The apical sixth ligand site is occupied by a chloride ligand as anticipated from the ESI-MS results. The observed Fe–OC(O)R bond distances in compound 3(b)-Cl are somewhat longer than those observed in other end-on iron(II)–carboxylate complexes (Table 1).49–56 We attribute this elongation to the steric constraints enforced by the chelating ligand, and the effect of having three anionic donors that produce a relatively electron-rich iron(II) center, thus weakening the Fe–OC(O)R bonds. Similar bond elongation was observed in [FeII(qn)2(Cl)]− (Table 1, qn = quinaldate), a carboxylate-rich iron(II) complex that also contains three anionic donors. Indeed, later in this report, we demonstrate substitution of the anionic chloride ligand by a neutral N-donor results in contraction of the Fe–OC(O)R bonds.
 | ||
| Fig. 2 ORTEP plot of the Ph4P+ salt of 3(b)-Cl (50% probability level). Hydrogen atoms, cation, and solvent molecules were omitted for clarity. | ||
| Fe–N1 | Fe–N2 | Fe–N3 | Fe–O1 | Fe–O2 | Fe–O/Cl | |
|---|---|---|---|---|---|---|
| a EXAFS-measured distances; all others obtained using X-ray diffraction techniques. b Ligand abbreviations used: qn = quinaldate; Py = pyridine; AcO = acetate; Mes2ArCO2 = 2,6-dimesitylbenzoate; bdtbpza = bis(3,5-di-tert-butylpyrazol-1-yl)acetic acid. | ||||||
| 3(b)-Cl | 2.251(4) | 2.181(4) | 2.179(4) | 2.081(3) | 2.103(4) | 2.3176(15) |
| 3(b)-Cl a | 5 @ 2.15a | 1 @ 2.31a | ||||
| [FeII(qn)2(Cl)]b65 | 2.0413(11) | 2.0705(11) | 2.2788(5) | |||
| 3(b)-Me22Im | 4 @ 2.14, 2 @ 1.94a | — | ||||
| [FeII(Py)2(Mes2ArCO2)2]b52 | 1.961(2) | 1.980(2) | ||||
| [FeII(MeIm)2(Mes2ArCO2)2]b50,55 | 2.028(2) | 1.989(2) | ||||
| [FeII(Py)4(AcO)2]b49,66 | 2.031(4) | 2.031(4) | ||||
| [FeII(bdtbpza)Cl]2b54 | 2.046(3) | 2.320(4) | ||||
| 5(b) | 4 @ 1.98a | 1 @ 1.66a | ||||
| [FeIV(O)(N4Py)]2+32 | 2.033(8) | 1.964(5) | Fe–N′ −1.949(5) | 1.639(5) | ||
| [FeIV(O)(N4Py)]2+67 | 5 @ 1.98a | 1 @ 1.64a | ||||
| [FeIV(O)(TPA)(NCCH3)]2+29 | 4 @ 1.98a | 1 @ 1.65a | ||||
| [FeIV(O)(TPA)(O2CCF3)]+68 | 4 @ 1.98, 1 @ 2.19a | 1 @ 1.66a | ||||
Two factors facilitated the isolation and characterization of 3(b)-Cl: the introduction of the n-butyl tail and the use of FeCl2 as an iron source. The tail enhanced the solubility of the mononuclear complex in organic solvents, thus retarding precipitation and polymerization, while the chloride ligand from FeCl2 served as the 6th ligand and prevented polymer formation. Alternative iron(II) sources ([FeII(OTf)2(NCCH3)2] or [FeII(NCCH3)4](BF4)2) yielded insoluble polymeric materials. Similar observations have been made elsewhere.64,65 The structure of 3(b)-Cl, with two equatorial carboxylates cis to a labile ligand site, demonstrates that by careful ligand design complexes that act as structural mimics of the active site of mononuclear nonheme iron enzymes can be obtained (Scheme 1, Fig. 2).
Towards the generation of high-valent intermediates
In order to generate oxoiron(IV) complexes utilizing P2DA ligands, we exposed the [FeII(P2DA)] complexes to oxygen atom transfer (OAT) reagents. Exposure of an aqueous solution of complex 3(a) to NaIO4 at 4 °C resulted in a rapid (5 s) conversion to a new species 4(a), as monitored by UV-vis absorption spectroscopy. Similar results were observed when alternative OAT reagents were used (such as H2O2, 3-chloroperoxybenzoic acid (m-CPBA)). 3(a) exhibits a single low intensity absorption feature (Figure S5, Supporting Information†) at λmax = 400 nm (ε = 600 M−1 cm−1), while the oxidized product 4(a) exhibits a single absorption feature at λmax = 480 nm (ε = 150 M−1 cm−1). The product solution was EPR silent, ruling out the possibility that 4(a) was a mononuclear iron(III) compound. ESI-MS analysis of the product solution showed an intense peak at m/z = 765.1, corresponding to the (K[Fe2(O)(P2DA)2])+ ion (Figure S6, Supporting Information†). This formulation would suggest the formation of a μ-oxo-bridged diiron(III) complex. Previous reports have shown μ-oxo-bridged diiron(III) compounds to display low intensity electronic absorption features similar to those observed in 4(a).69,70 Crystals of complex 4(a) were obtained by slow crystallization from aqueous solution confirming the formation of μ-oxo-bridged diiron(III) complex 4(a) upon exposure of 3(a) to OAT reagents (Figure S7, Supporting Information†).Often the use of temperatures below 0 °C facilitates the isolation of transient oxoiron(IV) intermediates for thorough spectroscopic characterization. Complex 3(a) was soluble only in H2O, but not in common organic solvents. We therefore designed n-butyl-functionalized complex 3(b)-Cl to increase solubility in common organic solvents such as CH2Cl2 and CH3OH (melting point CH2Cl2 = −97 °C, CH3OH = −98 °C). Exposure of a CH2Cl2 solution of 3(b)-Cl to 2-(tert-butylsulfonyl)iodosylbenzene (2-(tBuSO2)C6H4IO, sArIO) at −95 °C did not yield an oxoiron(IV) complex. UV-vis analysis of the reaction showed a reaction profile very similar to that observed in the reaction between 3(a) and NaIO4. 3(b)-Cl possesses two electronic absorption features in the visible region (Fig. 3, black trace λmax = 400 nm (ε = 650 M−1cm−1), 510 nm (ε = 675 M−1cm−1)) that, upon addition of sArIO, converted to a single feature with λmax = 480 nm (ε = 150 M−1cm−1) assigned to 4(b) (Fig. 3, orange trace). No features in the near-IR region of the spectrum, characteristic of nonheme oxoiron(IV) complexes, were observed.25 ESI-MS analysis showed an intense peak consistent with 4(b) being a μ-oxo-bridged diiron(III) complex (m/z = 877.0, (K[FeIII2(O)(nBu-P2DA)2])+, Figure S8, Supporting Information†). Crystals of 4(b) were obtained from a CH2Cl2/Et2O solution (Figure S9, Supporting Information†), which confirmed the formation of a μ-oxo-bridged diiron(III) compound.
 | ||
| Fig. 3 UV-vis spectral changes observed in the reaction of 3(b)-Cl (1.0 mM, solid black trace) with three equiv. sArIO in CH2Cl2 at −95 °C yielding 4(b) (solid orange trace). | ||
In summary, initial investigations into the reaction between an OAT reagent and [FeII(P2DA)] complexes did not yield the desired two-electron oxidized oxoiron(IV) complexes, but instead gave rise to μ-oxo-bridged diiron(III) products. The isolated diiron(III) products were highly stable and could not be converted to higher valent iron products. We suspected that the oxoiron(IV) species were indeed formed upon exposure of 3(a/b) to an OAT reagent; however, the nascent oxoiron(IV) species rapidly reacted with residual iron(II) starting material yielding the μ-oxo-bridged diiron(III) complexes (Scheme 5). We therefore addressed methods to accelerate the formation of the oxoiron(IV) intermediate and to inhibit the reaction of transient oxoiron(IV) with residual iron(II) starting material.
 | ||
| Scheme 5 Hypothesized route towards μ-oxo-bridged diiron(III) products in the reaction between 3(b)-X and OAT reagents. | ||
Reaction of 3(b)-Cl with AgBF4, in CH3CN resulted in the formation of a silver mirror in the reaction vessel, indicating the iron(II) complex was oxidized by the silver(I) salt. Reaction of 3(b)-Cl with TlPF6, an alternative halide abstraction reagent, yielded a polymeric orange material that was insoluble in common organic solvents. FT-IR analysis of the formed materials showed broad features centered at ∼1620 and 1390 cm−1 which are typical of polymeric μ-carboxylato materials (as in 3(a)).63 We concluded that removal of the chloride ligand in 3(b)-Cl facilitates intermolecular carboxylate-metal binding, resulting in polymeric species.
We then investigated 3(b)-Cl towards halide substitution. Exposure of a CH2Cl2 solution of 3(b)-Cl at −95 °C to 10 equiv. of (nBu4N)OH in CH3OH resulted in a red shift in the visible absorption spectrum (Figure S10, Supporting Information†). ESI-MS of the resulting solution showed peaks corresponding to [FeII(OH)(nBu-P2DA)]− (3(b)-OH, m/z = 428.1, Figure S11, Supporting Information†) and no peaks corresponding to 3(b)-Cl. Reaction of 3(b)-OH with sArIO, however, did not yield oxoiron(IV) product, and again gave indications that μ-oxo-bridged diiron(III) compound 4(b) was forming. Neutral σ-donor ligands reacted with 3(b)-Cl at −95 °C in CH2Cl2 (Fig. 4, pyridine (Py), 1-methylimidazole (MeIm), 1,2-dimethylimidazole (Me2Im), 4-dimethylaminopyridine (DMAP)), yielded new species as monitored by electronic absorption spectroscopy (Figures S12, 7, and S13 respectively†). ESI-MS analysis of the 3(b)-N samples showed 3(b)-Cl was no longer present but we also did not obtain any indications as to the formulation of the iron(II) N-adduct probably because of their lack of charge. 1H NMR analysis of the Me2Im adduct showed significant differences from 3(b)-Cl with fourteen signals observed (versus ten for 3(b)-Cl, Figure S14†). The additional peaks could be associated with the presence of a single Me2Im ligand bound to the iron(II) center, with the nBu-P2DA ligand maintaining the same coordination mode. We therefore assigned the names 3(b)-Py, 3(b)-MeIm, 3(b)-Me22Im, and 3(b)-DMAP to the adducts.
 | ||
| Fig. 4 UV-vis analysis of the reaction between 3(b)-Cl (black trace, 1.4 mM) and 100 equiv. 1-methyimidazole at −95 °C yielding 3(b)-MeIm (solid red trace). | ||
To gain a greater understanding of the structural and electronic properties of the N-adducts of 3(b) we turned to X-ray absorption spectroscopy (XAS). Fig. 5 shows the X-ray absorption near edge structure (XANES) features of 3(b)-Cl and 3(b)-Me22Im with their respective parameters listed in Table S2†. The iron K-edge of 3(b)-Cl is downshifted by ca. 1 eV relative to that of 3(b)-Me22Im, suggesting that 3(b)-Cl has a lower ionization potential than 3(b)-Me22Im, as would be expected for the substitution of an anionic chloride by a neutral imidazole ligand. The XANES spectra of both 3(b)-Cl and 3(b)-Me22Im exhibit weak broad pre-edge features, assigned to 1s to 3d transitions, which provide information about the geometries and electronic properties of these two complexes. These pre-edge features are fitted with 3 peaks (Table S2 and Figure S20†) with total areas, widths, positions, and splitting patterns similar to those found for other 6-coordinate S = 2 iron(II) complexes.71,72 In general, pre-edge areas of octahedral S = 2 iron(II) complexes vary between 3 to 6 units.71,72 XANES analysis suggests that both complexes are 6-coordinate and the anionic Cl− ligand in 3(b)-Cl has been displaced by neutral Me2Im to yield 3(b)-Me22Im.
 | ||
Fig. 5 XANES spectra of 3(b)-Cl (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ), 3(b)-Me22Im ( ), 3(b)-Me22Im (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ), and 5(b) ( ), and 5(b) ( ). Inset: magnified pre-edge features. ). Inset: magnified pre-edge features. | ||
Extended X-ray absorption fine structure (EXAFS) spectra of 3(b)-Cl and 3(b)-Me22Im are shown in Fig. 6A and 6B, respectively. The differences in both the k-space and r′-space data for the two complexes reflect the presence of the heavier chloride scatterer in 3(b)-Cl. The first shell of 3(b)-Cl was best fit with a shell of 5 O/N scatterers at 2.15 Å, corresponding to the donors of the nBu-P2DA ligand, and a chloride scatterer at ∼2.3 Å (Table 1 and S3†), in agreement with the distances found for 3(b)-Cl by X-ray crystallography (Table 1). On the other hand, the inner shell of 3(b)-Me22Im was best fit with two subsets of O/N ligands at 1.95 and 2.14 Å, corresponding respectively to the O and N sub-shells that arise from the nBu-P2DA ligand. A chloride scatterer was not required. Two subshells of C scatterers were required to simulate the outer shell features of 3(b)-Me22Im, which are assigned to C atoms α to the N donors. The shorter Fe⋯C paths derive from the pyridine donors, as found also for 3(b)-Cl, while the longer Fe⋯C paths arise from the Me2Im ligand, which are not required to fit the 3(b)-Cl data. These fits clearly demonstrate that the chloride ligand in 3(b)-Cl has been replaced in 3(b)-Me22Im. The resolution of the 1.95-Å O subshell from the 2.15-Å N subshell in 3(b)-Me22Im suggests the shortening of its Fe–OC(O)R bonds relative to those of 3(b)-Cl, consistent with the loss of the negative charge of the complex upon the replacement of the anionic Cl ligand with neutral imidazole.
 | ||
| Fig. 6 Fourier transforms (°) and their best fits () obtained from the Fe K-edge EXAFS data for 3(b)-Cl (A), 3(b)-Me22Im (B), and 5(b) (C). Fit parameters are provided in Tables 3 and S2† in bold. See footnote of Table S3† for more information. | ||
Characterization of the oxoiron(IV) complex
The oxoiron(IV) species (5(b)) can be generated at −95 °C by exposure of 3(b)-Me22Im to 3 equiv. sArIO in CH2Cl2 or to 3 equiv. m-CPBA in CH3OH (Figures S14† and 7, respectively). As illustrated in Fig. 7, the conversion in CH3OH could be followed by the replacement of features at 380 and 490 nm associated with 3(b)-Me22Im with less intense bands from the product at 510, 770, and 920 nm. Isosbestic points were observed at 400 and 560 nm, suggesting a direct conversion from 3(b)-Me22Im to 5(b). The bands at 770 and 920 nm associated with 5(b) are characteristic of d–d transitions in S = 1 oxoiron(IV) complexes as determined by MCD studies.25,73 The positions of these features relative to those of other oxoiron(IV) complexes provide a good indication of the relative ligand field splitting generated by the supporting ligands. Fig. 8 and Table 2 compare the most intense near-IR bands of several [FeIV(O)(L)] complexes and show that they decrease in energy in the order L = N4Py > TPA/NCCH3 > TPA/O2CCF3 > nBu-P2DA > L8Py2 > TMC/NCCH3 ∼ TMC-Py, reflecting variations in the equatorial donors. The observed red shift of the d–d transitions in the series occurs as equatorial pyridines are progressively substituted by weaker field donors such as NCCH3, carboxylates, or tertiary amine. | ||
| Fig. 7 Progress of the reaction between 3(b)-Me22Im (red trace) and three equiv. m-CPBA at −95 °C in CH3OH (1.0 mM) to yield 5(b) (green trace) as monitored by UV-vis-near-IR spectroscopy. | ||
![Near-IR regions of the electronic absorption spectra of [FeIV(O)(L)] complexes. All spectra were normalized to exhibit a common absorbance intensity for ease of comparison.](/image/article/2012/SC/c2sc01044e/c2sc01044e-f8.gif) | ||
| Fig. 8 Near-IR regions of the electronic absorption spectra of [FeIV(O)(L)] complexes. All spectra were normalized to exhibit a common absorbance intensity for ease of comparison. | ||
| Complex | λ max (ε) (nm, M−1cm−1) | D (cm−1) | E/D | A x,y,z/gnβn (T) | ΔEQ (mm s−1) | η | δ (mm s−1) |
|---|---|---|---|---|---|---|---|
| 5(b) | 770 (220) | 27 (2) | 0 | −22, −22, −5 | 1.13 | 0.2 | 0.04 |
| [FeIV(O)(N4Py)]2+74 | 695 (400) | 22 (2) | 0 | −21, −21, −5 | 0.93 | 0 | −0.04 |
| [FeIV(O)(TPA)(NCCH3)]2+68 | 724 (300) | 28 (2) | 0 | −23.5, −23.5, −5 | 0.93 | 0.9 | 0.01 |
| [FeIV(O)(TPA)(O2CCF3)]+68 | 745 (300) | — | — | — | 0.92 | — | 0.02 |
| [FeIV(O)(L8Py2)(NCCH3)]2+75 | 790 (260) | — | — | — | 1.79 | — | 0.08 |
| [FeIV(O)(TMC)(NCCH3)]2+28 | 824 (400) | 29 (2) | 0 | −23, −18, −3 | 1.23 | 0.5 | 0.17 |
| [FeIV(O)(TMC-Py)]2+76 | 834 (260) | 29 (2) | 0.15 | −22.5, −22.5, −5 | 1.08 | −0.35 | 0.18 |
Based on the intensity of its 770-nm band, varying yields of 5(b) were obtained upon treatment of the 3(b)-N complexes (N = Py, MeIm, Me2Im, DMAP) with sArIO at −95 °C in CH2Cl2 at 1 mM concentrations. The extinction coefficient (ε) for this band was determined to be 220 M−1cm−1 by parallel UV-vis analysis of the Mössbauer sample (see below). A maximum yield of 90% oxoiron(IV) was obtained with 3(b)-Me22Im as the precursor (Figure S16†). Yields of 5(b) diminished in the order 3(b)-DMAP (75%) > 3(b)-MeIm (40%) > 3(b)-Py (5%) (Figures S17–S19, Supporting Information†), and no 5(b) was obtained from anionic complexes 3(b)-Cl and 3(b)-OH under the same conditions. This trend can be rationalized by Scheme 5 where the yield of 5(b) is controlled by a competition between its rate of formation and its rate of decay by reaction with residual starting material 3(b)-X to give μ-oxo-bridged diiron(III) compound 4(b). For the anionic complexes 3(b)-Cl and 3(b)-OH, no 5(b) was formed because the rate of 5(b) decay was accelerated by the lower FeIII/II redox potentials of the iron(II) precursors, as suggested by electrochemical measurements (Figure S15†) and their lower K-edges (Fig. 5). However, the use of Me2Im or DMAP as an ancillary ligand provided the appropriate balance between the rates of formation and decay to allow 5(b) to be formed in significant yield for detailed spectroscopic characterization.
We attempted to acquire evidence for the formulation of 5(b) using ESI-MS, but did not observe any mass peaks that could be assigned. Similarly, we were unable to observe the νFeO of 5(b) using resonance Raman or FT-IR spectroscopies, probably due to the thermal and photo instability of 5(b). However our ability to prepare samples of 5(b) in CH3OH solution (Fig. 7), with m-CPBA as oxidant instead of sArIO in CH2Cl2, allowed us to characterize 5(b) by Mössbauer and XAS methods, which would have been much more difficult to accomplish for samples prepared in CH2Cl2.
Fig. 9 shows Mössbauer spectra of the sample containing 5(b) in frozen methanol solution. In zero field at a temperature of 4.2 K, the major feature is a quadrupole doublet with δ = 0.04 mm s−1 and ΔEQ = 1.13 mm s−1 that represented 70% of total iron in the sample. Subsequent optimization of reaction conditions allowed us to obtain 5(b) in yields as high as 90% according to UV-vis measurements. The value of δ is characteristic of the many examples of S = 1 oxoiron(IV) complexes reported to date, particularly those with pyridine ligands.24,25 The δ values in the [FeIV(O)(L)] series increase with the red shift of the corresponding near-IR bands (Fig. 8 and Table 2), in line with the progressive replacement of pyridine with weaker donors such as NCCH3, carboxylate, or tertiary amine.
 | ||
| Fig. 9 Mössbauer spectra (black hashed line) of 5(b) in frozen methanol solution measured between 4.2 K and 100 K with parallel applied magnetic fields indicated in the figure. The red solid lines are the spectral simulations using the parameters listed in Table 2. The black arrows highlight the spectral features belonging to the 10% diferric impurities (see text). In 4.2 K, 4 T and 8 T spectra, 20% of mononuclear high-spin ferric impurities was removed from the baseline. Additional spectra are listed in the SI†. | ||
Mössbauer spectra of 5(b) in applied magnetic fields were analyzed using the S = 1 spin Hamiltonian
 | (1) |
In addition to the major S = 1 oxoiron(IV) species, the spectra in Fig. 9 contain a diamagnetic species (10% of Fe) with δ = 0.46 mm s−1 and ΔEQ = −1.65 mm s−1. The isomer shift and quadrupole splitting indicate that this minority species is most likely the (μ-oxo)diiron(III) complex 4(b) suggested in Scheme 5. The rest of the iron in the sample, representing 20% of the total Fe, is in the form of a mononuclear high-spin iron(III) species (Figure S34†).
To gain insight into the structural properties of 5(b) we utilized XAS. XANES spectra of 5(b) and related complexes are depicted in Fig. 10. The spectrum of 5(b) exhibits an intense symmetric peak at 7114.3 eV with an area of 23.9 units, similar to those found for other S = 1 oxoiron(IV) complexes (Table S2†, Fig. 10),68,79 indicating that the introduction of two anionic carboxylate donors does not significantly affect the overall distortion from electronic centrosymmetry at the iron center. However the iron K-edge does downshift by approximately 1 eV with respect to previously reported oxoiron(IV) compounds (as marked with the arrow in Fig. 10), reflecting a lower ionization potential for the neutral oxoiron(IV) complex relative to its cationic analogs.
![XANES features of [FeIV(O)(N4Py)]2+ (blue),67 [FeIV(O)(TPA)(NCCH3)]2+ (pink),29 [FeIV(O)(TPA)(O2CCF3)]+ (green),68 and 5(b) (red). Inset: magnified pre-edge features.](/image/article/2012/SC/c2sc01044e/c2sc01044e-f10.gif) | ||
| Fig. 10 XANES features of [FeIV(O)(N4Py)]2+ (blue),67 [FeIV(O)(TPA)(NCCH3)]2+ (pink),29 [FeIV(O)(TPA)(O2CCF3)]+ (green),68 and 5(b) (red). Inset: magnified pre-edge features. | ||
The EXAFS data obtained for 5(b) is depicted in Fig. 6(C). Fitting the inner shell feature of 5(b) requires two subshells of Fe–O/N paths (Fits 13–17), where the inclusion of a Fe–O path of ∼1.66 Å significantly enhances the fit quality (Fits 16 and 17). The addition of two outer shell Fe–C paths at ∼2.8–3.0 Å, which are usually observed for mononuclear oxoiron(IV) complexes with pyridine donors,29,67,68 improves the fit quality as well (Fits 18–21). The inclusion of a Fe–N/O scatterer at ∼2.2 Å to Fit 20 (Fit 21) results in little change in the other parameters, but increases the F′ value, so such a path is not necessary. Thus, Fit 20 is the best fit for 5(b), which clearly has an FeO bond of 1.66 Å.
DFT calculations
DFT calculations were performed on 5(b) to gain further insights into the geometric and electronic structures of this complex. Apart from the obvious metric differences associated with the different oxidation states, geometry optimization of 5(b) in the S = 1 spin state (Fig. 11) shows a structure similar to that obtained crystallographically for 3(b)-Cl (Fig. 2), with metal–ligand bond lengths of 1.65 Å for the FeO bond, 1.92 Å for the Fe–OC(O)R bonds, 1.98 Å for the Fe–NPy bonds, and 2.12 Å for the axial Fe–Namine bond. These values are consistent with the EXAFS-derived results (Table 3). On the other hand, geometry optimization on the S = 2 spin state gave equatorial metal–ligand bond lengths that were dramatically elongated compared to those in the S = 1 spin state (Table S11†). Taken together, the DFT results support the assignment of 5(b) as an S = 1 oxoiron(IV) complex.
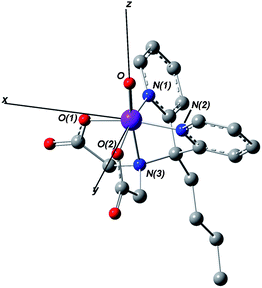 | ||
| Fig. 11 Geometry optimized structure of 5(b) in the gas phase obtained from DFT calculations for the S = 1 ground state. Hydrogen atoms were omitted for clarity. | ||
| Fit | FeO |
Fe–O/N | Fe–N/O | FeC |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| # | N | R | σ 2 | N | R | σ 2 | N | R | σ 2 | N | R | σ 2 | F | F′ |
| a k range = 2–12.4 Å−1, resolution ∼0.15 Å, back transform range 0.85–3.1 Å. N = 104, nidp = 14.7. See footnote of Table S3† for more information. | ||||||||||||||
| 13 | 6 | 1.99 | 13.8 | 142 | 0.1169 | |||||||||
| 14 | 5 | 1.99 | 10.0 | 111 | 0.0911 | |||||||||
| 15 | 4 | 2.00 | 6.0 | 1 | 2.23 | 1.9 | 75 | 0.0740 | ||||||
| 16 | 0.9 | 1.66 | 4.8 | 4 | 1.97 | 6.6 | 38 | 0.0377 | ||||||
| 17 | 0.9 | 1.67 | 5.3 | 4 | 1.97 | 4.9 | 1 | 2.16 | 2.4 | 31 | 0.0382 | |||
| 18 | 5 | 1.99 | 9.8 | 2 | 2.81 | 1.9 | 98 | 0.1223 | ||||||
| 3 | 3.00 | 1.0 | ||||||||||||
| 19 | 4 | 2.00 | 6.3 | 1 | 2.25 | 3.8 | 2 | 2.79 | 2.5 | 60 | 0.1011 | |||
| 3 | 3.00 | 1.4 | ||||||||||||
| 20 | 0.9 | 1.66 | 5.0 | 4 | 1.98 | 6.6 | 2 | 2.78 | 3.2 | 17 | 0.0295 | |||
| 3 | 2.99 | 2.4 | ||||||||||||
| 21 | 0.9 | 1.66 | 5.4 | 4 | 1.97 | 5.7 | 1 | 2.17 | 8.4 | 2 | 2.78 | 4.4 | 18 | 0.0468 |
| 3 | 2.99 | 2.7 | ||||||||||||
The DFT analysis of the S = 1 spin state of 5(b) allowed us to calculate the zero-field splitting (ZFS) parameter D, which originates from spin–orbit coupling between the ground state and the excited states within the S = 1 manifold as well as with excited S = 2 and S = 0 states.77 Following the same treatment as presented elsewhere (see Supporting Information†),78 we estimated D = 30.5 and 23.2 cm−1 for 5(b) and [FeIV(O)(N4Py)]2+, respectively, in good agreement with the values of 27 ± 2 and 22 ± 2 cm−1, respectively, determined experimentally by Mössbauer spectroscopy (Table 2). The DFT-calculated contributions from various spin states for 5(b) are DS−0 = 7.8 cm−1, DS−1 = 7.7 cm−1, and DS−2 = 15 cm−1, and for [FeIV(O)(N4Py)]2+ are DS−0 = 7.9 cm−1, DS−1 = 6.8 cm−1, and DS−2 = 8.5 cm−1. The much larger S = 2 spin state contribution to the ZFS of 5(b) compared to that of [FeIV(O)(N4Py)]2+ (15 vs. 8.5 cm−1) reflects a smaller vertical energy gap between the minimum of the S = 1 ground state potential energy surface (PES) and the S = 2 excited state PES of 5(b) (3550 cm−1 as calculated by DFT) compared to that of [FeIV(O)(N4Py)]2+ (6210 cm−1). To further explore the relative energies of the PES minima of the S = 1 and 2 states, we compared the DFT calculated energies based on the optimized structures of both spin states of 5(b) and [FeIV(O)(N4Py)]2+. For 5(b), the calculated electronic energy of the S = 1 spin state is ∼190 cm−1 lower than that of the optimized S = 2 spin state. By taking into account the corrections of zero-point, thermal, enthalpic and entropic energies, as well as the solvation energy calculated using the polarizable continuum model (PCM) on the optimized structures of both spin states obtained by the gas-phase calculations, this energy gap becomes ∼650 cm−1 with the S = 1 spin state still being the ground state. For [FeIV(O)(N4Py)]2+, the same calculation gave the free energy differences between the S = 1 and 2 spin states to be 2460 cm−1 with the S = 1 state being the ground state (Table S12†). These results indicate that the replacement of two pyridine donors of N4Py with carboxylates in the nBu-P2DA ligand results in a smaller energy splitting between the dxy and dx2−y2 orbitals that modulates the ground spin state between S = 1 and S = 2 (see below).
The DFT-calculated Mössbauer parameters provide additional support for the S = 1 ground state of 5(b). The parameters for the optimized S = 1 spin state, namely δ = 0.09 (0.04) mm s−1, ΔEQ = 1.10 (1.13) mm s−1 and η = 0.3 (0), are in good agreement with the experimental values listed in parentheses for comparison. In contrast, the calculated values for the optimized S = 2 spin state (δ = 0.25 mm s−1, ΔEQ = 0.45 mm s−1 and η = 0.7) deviate significantly from the experimental values, especially the isomer shift, which should satisfy the condition |δDFT − δexp| < 0.08 mm s−1 based on our DFT experience with oxoiron(IV) complexes supported by nitrogen rich ligands. Figure S35† shows that there is an excellent correlation between the observed and calculated isomer shifts of a series of S = 1 oxoiron(IV) complexes, for which the isomer shift becomes more positive with a decreasing number of pyridine donors in the equatorial plane perpendicular to the FeO bond. This observation can be further illustrated by an empirical correlation between the observed isomer shifts and the number of equatorial pyridine donors in S = 1 oxoiron(IV) complexes shown in Fig. 12. With one exception, the substitution of an equatorial pyridine donor by a weaker field ligand, such as carboxylate, CH3CN, or tertiary amine, generally increases the isomer shift by ∼0.05 mm s−1. Thus, going from [FeIV(O)(N4Py)]2+ (4 equatorial pyridines) to [FeIV(O)(TMC)(X)]2+ (X = NCCH3, Py) (0 equatorial pyridine) increases the isomer shift by ∼0.2 mm s−1. That the isomer shift of 5(b) lies in the middle of these two extremes reflects the equatorial donor strength of 5(b) and is consistent with its having two pyridines and two carboxylate ligands in the equatorial plane.
![Correlation between experimentally observed isomer shifts and number of equatorial pyridine ligands for a series of [FeIV(O)(L)] complexes. The correlation coefficient R2 is 0.87. Complex 5(b) is indicated with a solid dot. TMC-Py = 1-(2-pyridylmethyl)-4,8,11-trimethyl-1,4,8,11-tetraazacyclotetradecane; Me,HPytacn = 1-(2′-dipyridylmethyl)-4,7-dimethyl-1,4,7-triazacyclononane; L8Py2 = N,N′-bis(2-pyridylmethyl)-1,5-diazacyclooctane; BPMCN = N, N′-bis(2-pyridylmethyl)-N,N′-dimethyl-trans-1,2-diaminocyclohexane; BisPi = bispidine (2,4-bis(2-pyridyl)-3-(2-pyridylmethyl)-7-methyl-3,7-diazabicyclo[3.3.1]nonanone); Bn-TPEN = N-benzyl-N,N′,N′′-tris(2-pyridylmethyl)-1,2-diaminoethane.](/image/article/2012/SC/c2sc01044e/c2sc01044e-f12.gif) | ||
| Fig. 12 Correlation between experimentally observed isomer shifts and number of equatorial pyridine ligands for a series of [FeIV(O)(L)] complexes. The correlation coefficient R2 is 0.87. Complex 5(b) is indicated with a solid dot. TMC-Py = 1-(2-pyridylmethyl)-4,8,11-trimethyl-1,4,8,11-tetraazacyclotetradecane; Me,HPytacn = 1-(2′-dipyridylmethyl)-4,7-dimethyl-1,4,7-triazacyclononane; L8Py2 = N,N′-bis(2-pyridylmethyl)-1,5-diazacyclooctane; BPMCN = N, N′-bis(2-pyridylmethyl)-N,N′-dimethyl-trans-1,2-diaminocyclohexane; BisPi = bispidine (2,4-bis(2-pyridyl)-3-(2-pyridylmethyl)-7-methyl-3,7-diazabicyclo[3.3.1]nonanone); Bn-TPEN = N-benzyl-N,N′,N′′-tris(2-pyridylmethyl)-1,2-diaminoethane. | ||
To gain further insight into the electronic structure of 5(b), we also considered its near-IR spectral features. TD-DFT calculations (Figure S36 and Table S16†) nicely reproduced the relative shifts of the near-IR features observed in Fig. 8 and listed in Table 2. According to the calculations, the most intense feature can be assigned to the dxz/yz → dx2−y2 transition, and its low energy shoulder can be assigned to dxy → dx2−y2 and dxy → dxz/yz transitions, consistent with previous studies.42,73Fig. 13 shows a linear correlation between the observed and calculated λmax values in the near-IR region, which illustrates the dependence of the dx2−y2 orbital energy on the equatorial donor strength. As the dx2−y2 orbital is mainly involved in a σ-antibonding interaction with the equatorial ligands in these S = 1 oxoiron(IV) complexes, the presence of weaker carboxylate donors in 5(b) results in a dx2−y2 orbital of lower energy and hence gives rise to lower d–d transition energies (Fig. 14 and Table S16†). At the same time, the dxy orbital, which is largely nonbonding in [FeIV(O)(N4Py)]2+ (∼70% iron character for β-spin), has substantial π-antibonding interactions with the p orbitals of the equatorial O donors from the carboxylate ligands in 5(b) (∼50% iron character, and 36% O character from carboxylate ligands for β-spin) that destabilize this orbital (Fig. 14). Consequently, the introduction of carboxylate ligands to the equatorial plane of oxoiron(IV) complexes results in a decreased dxy/dx2−y2 orbital energy gap (Table S16†) and lowers the S = 2 excited state energy.
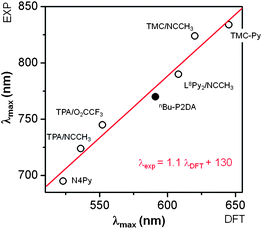 | ||
| Fig. 13 Correlation between the experimentally observed (y-axis) and DFT calculated (x-axis) λmax in the near-IR region of S = 1 oxoiron(IV) complexes with different supporting ligands. The correlation coefficient R2 is 0.99. Complex 5(b) is indicated with a solid dot. | ||
![Left: Spin-allowed d–d excitation energies obtained by TD-DFT for [FeIV(O)(N4Py)]2+ and 5(b) (the transition dxy → dz2 for 5(b) is strongly mixed with other transitions and has not been indicated). Right: isosurface plots of the 3d-type orbitals for the β-spin for 5(b).](/image/article/2012/SC/c2sc01044e/c2sc01044e-f14.gif) | ||
| Fig. 14 Left: Spin-allowed d–d excitation energies obtained by TD-DFT for [FeIV(O)(N4Py)]2+ and 5(b) (the transition dxy → dz2 for 5(b) is strongly mixed with other transitions and has not been indicated). Right: isosurface plots of the 3d-type orbitals for the β-spin for 5(b). | ||
In summary, DFT calculations confirm that complex 5(b) has an S = 1 ground state and a low-lying S = 2 excited state, resembling the electronic structures of other S = 1 oxoiron(IV) complexes.46 While the introduction of two carboxylate ligands reduces the ligand field splitting between iron dxy and dx2−y2 orbitals, the resulting energy gap is still too large to make the S = 2 state the ground state of 5(b). Clearly further adjustments on the equatorial ligands will be required to obtain a carboxylate-rich oxoiron(IV) complex with an S = 2 ground state.
Reactivity studies
At temperatures above −60 °C, 5(b) decayed at rates such that accurate kinetic analysis of its reactivity towards external substrates was unreliable using conventional methods. All kinetic measurements were therefore carried out at −60 °C using 5(b) prepared at −95 °C from 3(b)-Me22Im, because higher yields of 5(b) were obtained only when it was prepared at −95 °C. 5(b) has a half-life of 1200 s at −60 °C, decaying to yield an orange precipitate that was insoluble in MeOH or CH2Cl2. Post-decay analysis of the reaction mixture gave no indications of ligand oxidation products. The rate of decay of 5(b) was not dependent on the concentration of Me2Im (t1/2 = 1200 s, [Me2Im] = 0.05, 0.1, 0.25, 0.5 M, [5(b)] = 1 mM), suggesting that 5(b) does not oxidize Me2Im during its decay. However, the half-life of 5(b) nearly doubled when CD3OD or CH3OD was used as solvent in place of CH3OH (Table 4, kinetic isotope effect (KIE) = 1.7), indicating that MeOH, and specifically the alcohol functionality, is involved in the decay of 5(b). Given the strength of the O–H bond of MeOH, it is unlikely that MeOH is acting as a reductant of 5(b); however, the obtained KIE does suggest that either hydrogen bonding or a protonation event affects the rate-determining step in the decay of 5(b).| Complex | 5(b) | [FeIV(O)(N4Py)]2+ |
|---|---|---|
| a Half-lives and rate constants were determined at −60 °C in the presence of 500 equivalents Me2Im in MeOH. Spectral changes at λmax = 770 nm for 5(b) and 695 nm for [FeIV(O)(N4Py)]2+ were monitored. Half-lives and rate constants were averaged from at least 3 separate determinations, the standard deviation was <10% of the reported values. b DHT = 1-methyl-1,4-cyclohexadiene, P(Me)(Ph)2 = Methyldiphenylphosphine, Fc = ferrocene. | ||
| t 1/2 (s) | ||
| CH3OH | 1200 | 4000 |
| CD3OD | 2000 | |
| CH3OD | 2000 | |
| k 2 (M−1 s−1) | ||
| DHTb | 0.58 | 0.09 |
| P(Me)(Ph)2b | 2.8 | 0.7 |
| Fcb | 0.049 | 0.012 |
The 1200 s half-life of 5(b) at −60 °C provided us with ample time to investigate its reactivity towards readily oxidized substrates. In order to get a reliable measure of the relative potency of 5(b) as an oxidant, studies on the reactivity of [FeIV(O)(N4Py)]2+ under the same conditions were simultaneously carried out (Table 4). The oxidative reactivity of 5(b) for HAT at −60 °C was investigated with 1-methyl-1,4-cyclohexadiene (dihydrotoluene, DHT, C–H BDE = ∼77 kcal mol−1).80 DHT was chosen because of its miscibility with MeOH at −60 °C, in contrast to analogous substrates with comparably weak C–H bonds that are typically used for such studies, such as dihydroanthracene or 1,4-cyclohexadiene, which were immiscible with MeOH at −60 °C. Toluene was obtained in ∼35% yield and represented the only product found in the reaction mixtures, indicating that 5(b) acts as a HAT agent in this reaction and not as an OAT agent. ESI-MS analysis of the post-reaction mixture showed peaks corresponding to the potassium ion adducts of (μ-oxo)diiron(III) product 4(b) and monoiron(III) product [FeIII(OMe)(nBu-P2DA)], with no evidence for the formation of iron(II) products. Taken together, these results indicate that 5(b) effectively acts as a one-electron oxidant, with the 35% yield of toluene reflecting the conversion of ∼70% of oxoiron(IV) complex in the two-electron oxidation of DHT. Most importantly, 5(b) was found to have a second order rate constant (k2, Table 4) of 0.58 M−1 s−1 in the oxidation of DHT by HAT, which is 6-fold larger than that for [FeIV(O)(N4Py)]2+ (0.09 M−1 s−1, Figures S28 and S29†).
We also investigated the reactivities of 5(b) and [FeIV(O)(N4Py)]2+ with respect to OAT with a phosphine and electron transfer with ferrocene (Fc). In previous studies, rates of OAT reactions with nonheme oxoiron(IV) complexes were found to correlate directly with the electrophilicity of the oxoiron(IV) moiety.81,82 We thus anticipated that 5(b) would be a poorer OAT reagent than [FeIV(O)(N4Py)]2+, given the substitution of two neutral pyridine donors with negatively charged carboxylate ligands, making the oxo unit in 5(b) less electrophilic. Methyldiphenylphosphine (P(Me)(Ph)2) was chosen as a suitable phosphine as it showed good miscibility in MeOH at −60 °C, whereas various other triarylphosphines were poorly soluble in MeOH at −60 °C. Interestingly, the k2 value for the oxidation of P(Me)(Ph)2 by 5(b) was found to be 4-fold larger than that for [FeIV(O)(N4Py)]2+ (Table 4, Figures S30 and S31†), mirroring the results obtained in the oxidation of DHT. Methyldiphenylphosphine oxide (O = P(Me)(Ph)2) was the only product found in the reaction mixture in 90% yield, while UV-vis analysis of the post-reaction solution showed characteristic absorption features of 3(b)-Me22Im, confirming OAT from 5(b) yielding the 2-electron reduced product 3(b)-Me22Im. Similarly for the one-electron oxidation of Fc by 5(b) and [FeIV(O)(N4Py)]2+ to yield ferrocenium (Fc+) (Table 4), 5(b) was found to be a 4-fold better oxidant than [FeIV(O)(N4Py)]2+ (Figures S32 and S33†). Interestingly, the obtained k2 value for Fc oxidation by 5(b) was one order of magnitude smaller than that for DHT, suggesting that, while 5(b) is a viable electron transfer reagent, it is a superior HAT reagent. The obtained kinetic parameters for P(Me)(Ph)2 and Fc oxidation by 5(b) and [FeIV(O)(N4Py)]2+ clearly demonstrate that factors other than charge modulate the reactivity of oxoiron(IV) complexes. Thus in the three reactions investigated, 5(b) was found to be a faster oxidant than [FeIV(O)(N4Py)]2+ by approximately half an order of magnitude.
One approach, developed by Mayer, to understand the HAT reactivity of an oxoiron(IV) species is to consider the redox potential of the oxoiron(IV) moiety and the pKa of the hydroxoiron(III) product, which together define the strength of the FeIIIO–H bond formed in the reaction via the Bordwell–Polanyi relationship.47,48 The dicationic oxoiron complex [FeIV(O)(N4Py)]2+ is expected to have a higher redox potential than neutral 5(b), as evidenced by the lower Fe K-edge of 5(b) (Fig. 10) and results from DFT calculations; however, it is a less reactive HAT oxidant than 5(b). Therefore, the results obtained for 5(b) and [FeIV(O)(N4Py)]2+ cannot be rationalized based on the charge of the complex. Alternatively, it can be argued that the higher pKa of the FeIIIO–H unit induced by the anionic carboxylate donors makes the O–H bond of the hydroxoiron(III) product [FeIII(OH)(nBu-P2DA)] stronger than the O–H bond in [FeIII(OH)(N4Py)]2+. This relatively higher pKa value could then enhance the reactivity of 5(b) compared to [FeIV(O)(N4Py)]2+ and compensate for the lower relative redox potential of 5(b). A similar argument has been used in a previous report to rationalize the observation that electron-rich axial donors in [FeIV(O)(TMP˙)(p-Y-PyO)]+ (TMP˙ = meso-tetramesitylporphyrin radical; PyO = pyridine-N-oxide; Y = OMe, Me, H, Cl) enhanced the HAT reactivity of the oxoiron(IV) moiety.83 This counterintuitive trend was ascribed to the effect of electron-donating axial ligands in strengthening the FeIIIO–H bond, providing a greater driving force for HAT. The anionic carboxylate donors of 5(b) may have the same effect by increasing the pKa of the FeIIIO–H moiety in nBu-P2DA compared to N4Py, thereby strengthening the FeIIIO–H bond to provide a greater driving force for HAT.
The superior HAT reactivity of 5(b) could also be rationalized using Shaik's exchange-enhanced-reactivity (EER) model, which is often used to explain the HAT reactivity of S = 1 oxoiron(IV) complexes.40,46 In the EER model HAT reactivity of S = 1 oxoiron(IV) complexes occurs on the more reactive excited state (S = 2) surface, which is populated upon spin-crossover from the ground triplet S = 1 state to the quintet S = 2 surface along the reaction coordinate. The proximity of the triplet ground state to the quintet surface in any S = 1 oxoiron(IV) complex therefore has a strong influence on its reactivity. Both experiment (Mössbauer) and theory (DFT) provide us with important insights into the triplet/quintet energy difference in 5(b), showing that it has a lower lying quintet surface than [FeIV(O)(N4Py)]2+. Analysis of the Mössbauer data shows that 5(b) has a larger D value (27 cm−1) than [FeIV(O)(N4Py)]2+ (22 cm−1). As the magnitude of D is governed by the spin–orbit coupling between the ground S = 1 state and the various excited states,77,78 the larger D value reflects a smaller triplet/quintet separation for 5(b). The larger D value found experimentally for 5(b) is reproduced by DFT calculations and can be attributed to the weaker equatorial field exerted by the anionic carboxylate donors of 5(b). This weaker equatorial field decreases the triplet/quintet gap by ∼2700 cm−1 (Tables S12, S13†) with respect to [FeIV(O)(N4Py)]2+.
Besides the above-mentioned factors, steric effects may also play a role in the relative reactivities of 5(b) and [FeIV(O)(N4Py)]2+. A consideration of the frontier molecular orbitals (FMOs) of the S = 1 oxoiron(IV) unit suggests that the electrophilic attack of oxoiron(IV) on the substrate C–H bond mainly involves overlap between the π* MOs of the oxoiron(IV) unit and the target substrate C–H bond. Such an attack at the π* MOs ideally requires an FeO–HC angle of 120°.42,45,46 Based on a NRVS study of [FeIV(O)(N4Py)]2+,84 it was postulated by Solomon that the α-H-atoms on the pyridine ligands may act as a barrier for the π-approach of substrate C–H bonds towards the oxoiron(IV) unit of [FeIV(O)(N4Py)]2+. This partial shielding of the oxoiron(IV) center was conjectured to result in the retardation of the rate at which the electrophilic oxo can interact with C–H bonds. In 5(b) two of the four pyridine ligands of [FeIV(O)(N4Py)]2+ are replaced with carboxylate ligands, which should lower the steric barrier for a π-approach by substrate, as shown by the DFT-optimized structure of 5(b) (Fig. 11). On the other hand, consideration of the FMOs of the S = 2 oxoiron(IV) unit suggests that C–H bond attack would involve the σ* MO. Such a σ-approach would ideally require an Fe O–HC angle of 180°,42,45,46 and should not encounter any steric hindrance from either pentadentate ligand. Based on this steric comparison, we suggest that the reduced steric crowding of the π-approach pathway may allow 5(b) to be a more reactive HAT reagent than [FeIV(O)(N4Py)]2+.
While all three rationales discussed above have been used to explain the behavior of metal–oxo complexes in HAT reactions, the first two may not necessarily apply to OAT and electron transfer reactivity, as these reactions involve neither C–H bond cleavage nor O–H bond formation. As both OAT and electron transfer would be expected to be more facile at the more electrophilic oxo, we anticipated [FeIV(O)(N4Py)]2+ to be more reactive than 5(b). However, we observed the opposite trend where 5(b) was more reactive than [FeIV(O)(N4Py)]2+, which may be rationalized by the lower steric hindrance around the oxoiron(IV) center in 5(b) providing for easier substrate approach to the oxoiron(IV) unit.
From the data presented above, it is clear that more work needs to be done to assess the various factors that can affect the reactivity of the oxoiron(IV) unit. Systematic studies of oxoiron(IV) reactivity to date have focused mainly on complexes supported by either tetramethylcyclam81 or polydentate ligands with pyridine donors.30,85 Other donor groups should be investigated to elucidate the role equatorial donors play in modulating oxoiron(IV) reactivity. In an important recent study, Nam and co-workers report the synthesis and characterization of [FeIV(O)(Me3NTB)]2+ (Me3NTB = tris((N-methyl-benzimidazol-2-yl)methyl)amine), the first example of an oxoiron(IV) complex with benzimidazole donors.37 This complex exhibits the highest HAT reactivity thus far of any synthetic mononuclear oxoiron(IV) complex, with a k2 value for dihydroanthracene oxidation of 3100 M−1 s−1 at −40 °C, which is more than a thousand-fold faster than that for the corresponding 5-Me3TPA (5-Me3TPA = tris(5-methyl-2-pyridylmethyl)amine) complex (1.2 M−1 s−1).86 Despite the difference in heterocyclic N-donors (benzimidazole vs. pyridine respectively), both complexes have S = 1 ground states and comparable zero field splittings (D = 28 cm−1) that indicate similarly accessible S = 2 excited states for the two complexes. This comparison emphasizes the point that the nature of the donor ligands may be critical in controlling reactivity and more systematic studies are needed to gain further insight into this key question.
Conclusions
We have developed a novel carboxylate-rich pentadentate ligand (N-(1′,1′-(bis-(2-pyridyl)methyl)iminodiacetate), P2DA) that will bind iron(II) yielding a mononuclear octahedral complex with two carboxylate donors in the equatorial plane. This ligand has been designed to mimic the ligand sphere found in the active site of a superfamily of mononuclear non-heme iron enzymes. Generation of the carboxylate-rich oxoiron(IV) complex of nBu-P2DA was achieved by deactivation of the iron(II) precursor in order to prevent undesired side reactions between the oxoiron(IV) species and residual iron(II). The carboxylate donors in [FeIV(O)(nBu-P2DA)] reduce the redox potential of the iron center when compared to other charged synthetic oxoiron(IV) complexes. Despite the lower redox potential, the oxidative C–H bond activation reactivity of [FeIV(O)(nBu-P2DA)] is not diminished relative to previously synthesized oxoiron(IV) complexes and is in fact enhanced. This outcome may be the result of one of three factors or a combination of all three: a) a hydroxoiron(III) product stabilized by the anionic carboxylate donors, b) a smaller gap between the S = 1 ground state and the S = 2 excited state that enhances the reactivity of the oxoiron(IV) moiety, and c) lower steric bulk of the carboxylate donors in the complex allowing for more facile substrate access to the oxoiron(IV) moiety.[FeIV(O)(nBu-P2DA)] represents the closest structural mimic reported thus far of the active oxidant found in TauD and related α-ketoglutarate-dependent iron enzymes, the oxoiron(IV) species of which have two weak-field carboxylate donors in the plane perpendicular to the FeO bond. Although [FeIV(O)(nBu-P2DA)] has a similar coordination environment, the nBu-P2DA ligand does not exert a weak enough field to make the S = 2 spin state the ground state of the oxoiron(IV) center as found in TauD ‘J’. It would thus appear that the common facial N3 ligand set found in nBu-P2DA and N4Py exerts a sufficiently strong ligand field to make S = 1 the ground spin state for the oxoiron(IV) unit. As the DFT calculations indicate, the energy gap between the S = 1 and S = 2 configurations in 5(b) is only ∼650 cm−1, future endeavors will be focused on modifications to nBu-P2DA to generate carboxylate-rich S = 2 oxoiron(IV) complexes.
Acknowledgements
This work was supported by the National Institutes of Health (grants GM-33162 to L.Q. and EB001475 to E.M. and a post-doctoral fellowship GM-087895 to A.McD.) and the National Science Foundation (grants CHE1058248 to L.Q. and CHE070073 to E.L.B. through TeraGrid resources provided by the NCSA). The authors would like to thank Dr Victor G. Young, Jr. and Mr Greg Rohde of the University of Minnesota X-Ray Crystallographic Laboratory for the solution of the X-ray crystal structures of 3(b)-Cl, 4(a), and 4(b). We also thank Dr Erik R. Farquhar of the Center for Synchrotron Biosciences at Case Western Reserve University for mail-in XAS data collection for 3(b)-Cl, 3(b)-Me22Im, and 5(b) at beamline X3B of the National Synchrotron Light Source (Brookhaven National Laboratory). Operation of NSLS X3B is supported by NIH grant EB-009998. The NSLS is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-98CH10886.References
- R. H. Holm, P. Kennepohl and E. I. Solomon, Chem. Rev., 1996, 96, 2239–2314 CrossRef CAS.
- E. I. Solomon, T. C. Brunold, M. I. Davis, J. N. Kemsley, S.-K. Lee, N. Lehnert, F. Neese, A. J. Skulan, Y.-S. Yang and J. Zhou, Chem. Rev., 2000, 100, 235–349 CrossRef CAS.
- M. Costas, M. P. Mehn, M. P. Jensen and L. Que Jr, Chem. Rev., 2004, 104, 939–986 CrossRef CAS.
- E. L. Hegg and L. Que Jr, Eur. J. Biochem., 1997, 250, 625–629 CAS.
- L. Que, Nat. Struct. Biol., 2000, 7, 182–184 CrossRef CAS.
- K. D. Koehntop, J. P. Emerson and L. Que Jr, JBIC, J. Biol. Inorg. Chem., 2005, 10, 87–93 CrossRef CAS.
- J. M. Bollinger Jr, J. C. Price, L. M. Hoffart, E. W. Barr and C. Krebs, Eur. J. Inorg. Chem., 2005, 4245–4254 CrossRef.
- J. M. Bollinger Jr and C. Krebs, J. Inorg. Biochem., 2006, 100, 586–605 CrossRef.
- C. Krebs, D. Galonić Fujimori, C. T. Walsh and J. M. Bollinger Jr, Acc. Chem. Res., 2007, 40, 484–492 CrossRef CAS.
- J. C. Price, E. W. Barr, B. Tirupati, J. M. Bollinger Jr and C. Krebs, Biochemistry, 2003, 42, 7497–7508 CrossRef CAS.
- J. C. Price, E. W. Barr, T. E. Glass, C. Krebs and J. M. Bollinger Jr, J. Am. Chem. Soc., 2003, 125, 13008–13009 CrossRef CAS.
- P. J. Riggs-Gelasco, J. C. Price, R. B. Guyer, J. H. Brehm, E. W. Barr, J. M. Bollinger Jr and C. Krebs, J. Am. Chem. Soc., 2004, 126, 8108–8109 CrossRef CAS.
- J. C. Price, E. W. Barr, L. M. Hoffart, C. Krebs and J. M. Bollinger Jr, Biochemistry, 2005, 44, 8138–8147 CrossRef CAS.
- L. M. Hoffart, E. W. Barr, R. B. Guyer, J. M. Bollinger Jr and C. Krebs, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14738–14743 CrossRef CAS.
- D. P. Galonic, E. W. Barr, C. T. Walsh, J. M. Bollinger Jr and C. Krebs, Nat. Chem. Biol., 2007, 3, 113–116 CrossRef CAS.
- D. Galonic Fujimori, E. W. Barr, M. L. Matthews, G. M. Koch, J. R. Yonce, C. T. Walsh, J. M. Bollinger Jr, C. Krebs and P. J. Riggs-Gelasco, J. Am. Chem. Soc., 2007, 129, 13408–13409 CrossRef.
- B. E. Eser, E. W. Barr, P. A. Frantom, L. Saleh, J. M. Bollinger, C. Krebs and P. F. Fitzpatrick, J. Am. Chem. Soc., 2007, 129, 11334–11335 CrossRef CAS.
- P. L. Roach, I. J. Clifton, C. M. Hensgen, N. Shibata, C. J. Schofield, J. Hajdu and J. E. Baldwin, Nature, 1997, 387, 827–830 CrossRef CAS.
- Z. Zhang, J.-S. Ren, I. J. Clifton and C. J. Schofield, Chem. Biol., 2004, 11, 1383–1394 CrossRef CAS.
- N. I. Burzlaff, P. J. Rutledge, I. J. Clifton, C. M. H. Hensgens, M. Pickford, R. M. Adlington, P. L. Roach and J. E. Baldwin, Nature, 1999, 401, 721–724 CrossRef CAS.
- S. Sinnecker, N. Svensen, E. W. Barr, S. Ye, J. M. Bollinger, F. Neese and C. Krebs, J. Am. Chem. Soc., 2007, 129, 6168–6179 CrossRef CAS.
- O. A. Andersen, T. Flatmark and E. Hough, J. Mol. Biol., 2001, 34, 279–291 CrossRef.
- M. L. Neidig, C. D. Brown, K. M. Light, D. G. Fujimori, E. M. Nolan, J. C. Price, E. W. Barr, J. M. Bollinger Jr, C. Krebs, C. T. Walsh and E. I. Solomon, J. Am. Chem. Soc., 2007, 129, 14224–14231 CrossRef CAS.
- X. Shan and L. Que Jr, J. Inorg. Biochem., 2006, 100, 421–433 CrossRef CAS.
- L. Que Jr, Acc. Chem. Res., 2007, 40, 493–500 CrossRef.
- W. Nam, Acc. Chem. Res., 2007, 40, 522–531 CrossRef CAS.
- C. A. Grapperhaus, B. Mienert, E. Bill, T. Weyhermüller and K. Wieghardt, Inorg. Chem., 2000, 39, 5306–5317 CrossRef CAS.
- J.-U. Rohde, J.-H. In, M. H. Lim, W. W. Brennessel, M. R. Bukowski, A. Stubna, E. Münck, W. Nam and L. Que Jr, Science, 2003, 299, 1037–1039 CrossRef CAS.
- M. H. Lim, J.-U. Rohde, A. Stubna, M. R. Bukowski, M. Costas, R. Y. N. Ho, E. Münck, W. Nam and L. Que Jr, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3665–3670 CrossRef CAS.
- J. Kaizer, E. J. Klinker, N. Y. Oh, J.-U. Rohde, W. J. Song, A. Stubna, J. Kim, E. Münck, W. Nam and L. Que Jr, J. Am. Chem. Soc., 2004, 126, 472–473 CrossRef CAS.
- V. Balland, M.-F. Charlot, F. Banse, J.-J. Girerd, T. A. Mattioli, E. Bill, J.-F. Bartoli, P. Battioni and D. Mansuy, Eur. J. Inorg. Chem., 2004, 301–308 CrossRef CAS.
- E. J. Klinker, J. Kaizer, W. W. Brennessel, N. L. Woodrum, C. J. Cramer and L. Que Jr, Angew. Chem., Int. Ed., 2005, 44, 3690–3694 CrossRef CAS.
- J. Bautz, M. R. Bukowski, M. Kerscher, A. Stubna, P. Comba, A. Lienke, E. Münck and L. Que Jr, Angew. Chem., Int. Ed., 2006, 45, 5681–5684 CrossRef CAS.
- J. England, M. Martinho, E. R. Farquhar, J. R. Frisch, E. L. Bominaar, E. Münck and L. Que Jr, Angew. Chem., Int. Ed., 2009, 48, 3622–3626 CrossRef CAS.
- J. England, Y. Guo, E. R. Farquhar, V. G. Young Jr, E. Münck and L. Que Jr, J. Am. Chem. Soc., 2010, 132, 8635–8644 CrossRef CAS.
- D. C. Lacy, R. Gupta, K. L. Stone, J. Greaves, J. W. Ziller, M. P. Hendrich and A. S. Borovik, J. Am. Chem. Soc., 2010, 132, 12188–12190 CrossRef CAS.
- M. S. Seo, N. H. Kim, K.-B. Cho, J. E. So, S. K. Park, M. Clemancey, R. Garcia-Serres, J.-M. Latour, S. Shaik and W. Nam, Chem. Sci., 2011, 2, 1039–1045 RSC.
- S. Hong, Y.-M. Lee, K.-B. Cho, K. Sundaravel, J. Cho, M. J. Kim, W. Shin and W. Nam, J. Am. Chem. Soc., 2011, 133, 11876–11879 CrossRef CAS.
- H. Hirao, D. Kumar, L. Que Jr and S. Shaik, J. Am. Chem. Soc., 2006, 128, 8590–8606 CrossRef CAS.
- S. Shaik, H. Hirao and D. Kumar, Acc. Chem. Res., 2007, 40, 532–542 CrossRef CAS.
- L. Bernasconi, M. J. Louwerse and E. J. Baerends, Eur. J. Inorg. Chem., 2007, 3023–3033 CrossRef CAS.
- A. Decker, J.-U. Rohde, E. J. Klinker, S. D. Wong, L. Que Jr and E. I. Solomon, J. Am. Chem. Soc., 2007, 129, 15983–15996 CrossRef CAS.
- S. Ye and F. Neese, Curr. Opin. Chem. Biol., 2009, 13, 89–98 CrossRef CAS.
- D. Janardanan, Y. Wang, P. Schyman, L. Que Jr and S. Shaik, Angew. Chem., Int. Ed., 2010, 49, 3342–3346 CAS.
- C. Geng, S. Ye and F. Neese, Angew. Chem., Int. Ed., 2010, 49, 5717–5720 CrossRef CAS.
- S. Shaik, H. Chen and D. Janardanan, Nat. Chem., 2011, 3, 19–27 CrossRef CAS.
- J. M. Mayer, Acc. Chem. Res., 1998, 31, 441–450 CrossRef CAS.
- J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS.
- B. Singh, J. R. Long, F. F. deBiani, D. Gatteschi and P. Stavropoulos, J. Am. Chem. Soc., 1997, 119, 7030–7047 CrossRef CAS.
- J. R. Hagadorn, L. Que Jr and W. B. Tolman, J. Am. Chem. Soc., 1998, 120, 13531–13532 CrossRef CAS.
- J. R. Hagadorn, L. Que Jr, W. B. Tolman, I. Prisecaru and E. Münck, J. Am. Chem. Soc., 1999, 121, 9760–9761 CrossRef CAS.
- J. R. Hagadorn, L. Que Jr and W. B. Tolman, Inorg. Chem., 2000, 39, 6086–6090 CrossRef CAS.
- A. Beck, B. Weibert and N. I. Burzlaff, Eur. J. Inorg. Chem., 2001, 521–527 CrossRef CAS.
- A. Beck, A. Barth, E. Hulbner and N. Burzlaff, Inorg. Chem., 2003, 42, 7182–7188 CrossRef CAS.
- S. J. Friese, B. E. Kucera, L. Que and W. B. Tolman, Inorg. Chem., 2006, 45, 8003–8005 CrossRef CAS.
- P. C. A. Bruijnincx, M. Lutz, A. L. Spek, W. R. Hagen, B. M. Weckhuysen, G. van Koten and R. J. M. Klein Gebbink, J. Am. Chem. Soc., 2007, 129, 2275–2286 CrossRef CAS.
- S. Friedle, E. Reisner and S. J. Lippard, Chem. Soc. Rev., 2010, 39, 2768–2779 RSC.
- M. Renz, C. Hemmert and B. Meunier, Eur. J. Org. Chem., 1998, 1271–1273 CrossRef CAS.
- P. J. Arnold, S. C. Davies, J. R. Dilworth, M. C. Durrant, D. V. Griffiths, D. L. Hughes, R. L. Richards and P. C. Sharpe, J. Chem. Soc., Dalton Trans., 2001, 736–746 RSC.
- Y. Zang, J. Kim, Y. Dong, E. C. Wilkinson, E. H. Appelman and L. Que Jr, J. Am. Chem. Soc., 1997, 119, 4197–4205 CrossRef CAS.
- G. Roelfes, M. Lubben, K. Chen, R. Y. N. Ho, A. Meetsma, S. Genseberger, R. M. Hermant, R. Hage, S. K. Mandal, V. G. Young Jr, Y. Zang, H. Kooijman, A. L. Spek, L. Que Jr and B. L. Feringa, Inorg. Chem., 1999, 38, 1929–1936 CrossRef CAS.
- L.-J. Ming, in Physical Methods in Bioinorganic Chemistry. Spectroscopy and Magnetism, ed. L. Que Jr, University Science Books, Sausalito, CA, 2000, pp. 375–464 Search PubMed.
- G. B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227–250 CrossRef CAS.
- Y.-F. Song, J. F. Berry, E. Bill, E. Bothe, T. Weyhermüller and K. Wieghardt, Inorg. Chem., 2007, 46, 2208–2219 CrossRef CAS.
- D. T. Houghton, N. W. Gydesen, N. Arulsamy and M. P. Mehn, Inorg. Chem., 2009, 49, 879–887 CrossRef.
- B. Singh, J. R. Long, G. C. Papaefthymiou and P. Stavropoulos, J. Am. Chem. Soc., 1996, 118, 5824–5825 CrossRef CAS.
- J.-U. Rohde, S. Torelli, X. Shan, M. H. Lim, E. J. Klinker, J. Kaizer, K. Chen, W. Nam and L. Que Jr, J. Am. Chem. Soc., 2004, 126, 16750–16761 CrossRef CAS.
- J.-U. Rohde, A. Stubna, E. L. Bominaar, E. Münck, W. Nam and L. Que Jr, Inorg. Chem., 2006, 45, 6435–6445 CrossRef CAS.
- H. Zheng, Y. Zang, Y. Dong, V. G. Young Jr and L. Que Jr, J. Am. Chem. Soc., 1999, 121, 2226–2235 CrossRef CAS.
- A. Mairata i Payeras, R. Y. N. Ho, M. Fujita and L. Que Jr, Chem.–Eur. J., 2004, 10, 4944–4953 CrossRef CAS.
- C. R. Randall, L. Shu, Y.-M. Chiou, K. S. Hagen, M. Ito, N. Kitajima, R. J. Lachicotte, Y. Zang and L. Que Jr, Inorg. Chem., 1995, 34, 1036–1039 CrossRef CAS.
- T. E. Westre, P. Kennepohl, J. G. DeWitt, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1997, 119, 6297–6314 CrossRef CAS.
- A. Decker, J.-U. Rohde, L. Que Jr and E. I. Solomon, J. Am. Chem. Soc., 2004, 126, 5378–5379 CrossRef CAS.
- E. J. Klinker, T. A. Jackson, M. P. Jensen, A. Stubna, G. Juhász, E. L. Bominaar, E. Münck and L. Que Jr, Angew. Chem., Int. Ed., 2006, 45, 7394–7397 CrossRef CAS.
- Y. Zhou, X. Shan, R. Mas-Ballesté, M. R. Bukowski, A. Stubna, M. Chakrabarti, L. Slominski, J. A. Halfen, E. Münck and L. Que Jr, Angew. Chem., Int. Ed., 2008, 47, 1896–1899 CrossRef CAS.
- A. Thibon, J. England, M. Martinho, V. G. Young Jr, J. R. Frisch, R. Guillot, J.-J. Girerd, E. Münck, L. Que Jr and F. Banse, Angew. Chem., Int. Ed., 2008, 47, 7064–7067 CrossRef CAS.
- F. Neese and E. I. Solomon, Inorg. Chem., 1998, 37, 6568–6582 CrossRef CAS.
- A. Chanda, X. Shan, M. Chakrabarti, W. C. Ellis, D. L. Popescu, F. Tiago de Oliveira, D. Wang, L. Que Jr, T. J. Collins, E. Münck and E. L. Bominaar, Inorg. Chem., 2008, 47, 3669–3678 CrossRef CAS.
- T. A. Jackson, J.-U. Rohde, M. S. Seo, C. V. Sastri, R. DeHont, T. Ohta, T. Kitagawa, E. Münck, W. Nam and L. Que Jr, J. Am. Chem. Soc., 2008, 130, 12394–12407 CrossRef CAS.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, Taylor & Francis, Boca Raton, 2007 Search PubMed.
- C. V. Sastri, J. Lee, K. Oh, Y. J. Lee, J. Lee, T. A. Jackson, K. Ray, H. Hirao, W. Shin, J. A. Halfen, J. Kim, L. Que Jr, S. Shaik and W. Nam, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 19181–19186 CrossRef CAS.
- S. N. Dhuri, M. S. Seo, Y.-M. Lee, H. Hirao, Y. Wang, W. Nam and S. Shaik, Angew. Chem., Int. Ed., 2008, 47, 3356–3359 CrossRef CAS.
- Y. Kang, H. Chen, Y. J. Jeong, W. Lai, E. H. Bae, S. Shaik and W. Nam, Chem.–Eur. J., 2009, 15, 10039–10046 CrossRef CAS.
- S. D. Wong, C. B. Bell, L. V. Liu, Y. Kwak, J. England, E. E. Alp, J. Zhao, L. Que and E. I. Solomon, Angew. Chem., Int. Ed., 2011, 50, 3215–3218 CrossRef CAS.
- E. J. Klinker, S. Shaik, H. Hirao and L. Que Jr, Angew. Chem., Int. Ed., 2009, 48, 1291–1295 CrossRef CAS.
- G. Xue, R. De Hont, E. Münck and L. Que Jr., Nat. Chem., 2010, 2, 400–405 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: a description of all methods. Full experimental details for the synthesis of compounds 1–4(a,b), 3(b)-X and 5(b) and the reactivity studies. FT-IR and ESI-MS spectra for compounds 3–4(a,b), X-ray diffraction methods and data for 3(b)-Cl and 4(a,b). Fe K-edge XANES and EXAFS data and fitting analyses. UV-vis spectra for the synthesis of 3(b)-X and their conversion to 5(b). Mössbauer analysis of residual FeIII products found in the synthesis of 5(b). Details of DFT and TD-DFT computations for the analyses of 5(b) and related compounds. CCDC reference numbers 857970–857972. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2sc01044e |
| This journal is © The Royal Society of Chemistry 2012 |