Streamlined access to conjugation-ready glycans by automated synthesis†
Lenz
Kröck
ab,
Davide
Esposito
ab,
Bastien
Castagner‡
,
Cheng-Chung
Wang§
ab,
Pascal
Bindschädler¶
ab and
Peter H.
Seeberger
*ab
aMax-Planck-Institute of Colloids and Interfaces, Department of Biomolecular Systems, Am Mühlenberg 1, 14476 Potsdam, Germany. E-mail: Peter.Seeberger@mpikg.mpg.de
bFreie Universität Berlin, Institute of Chemistry and Biochemistry, Arnimallee 22, 14195 Berlin, Germany
First published on 20th February 2012
Abstract
Understanding the structure–function relationship of carbohydrates in biological systems is a major challenge. Such investigations require fast and reliable access to structurally-defined oligosaccharides. To date, oligosaccharide synthesis has been considered technically difficult and only parts of the synthetic process have been automated using solution and solid-phase techniques. Here, we describe a versatile platform integrating a new synthesis strategy and a fully-automated oligosaccharide synthesizer. Structurally diverse conjugation-ready oligosaccharides can be generated for the creation of glycoconjugates and microarrays. Biologically-significant oligosaccharides of increasing length, structural complexity, and chemical diversity were produced, including glycans found on the surfaces of pathogenic bacteria, and those with integral roles in inflammatory and immune responses.
Introduction
Analysis of the glycome has revealed the vast structural diversity and complexity of carbohydrates. However, for researchers and clinicians to fully appreciate the roles played by these macromolecules in biological systems, the structure–function relationship of carbohydrates must be established. Progress on this front has been impeded by the technical difficulty of synthesizing carbohydrates. By comparison, well-defined oligopeptides and oligonucleotides have been widely available for some time due to fully-automated solid-phase synthesis methods, enabling researchers to access desired oligomers and exploit diagnostic and therapeutic opportunities.1,2 The characteristics of carbohydrate architecture make it impossible for fully automated oligosaccharide synthesis to be achieved by a simple modification of oligopeptide and oligonucleotide synthesis methodologies. Unlike proteins and nucleic acids, oligosaccharides are not merely linear chains, but rather, are branched structures (Fig. 1A). Consequently, there are unique challenges integral to the synthesis of carbohydrates. An obvious consideration is the anomeric carbon at the linkage between two monomers, which constitutes a stereogenic center. Regulating the stereochemical outcome of glycoside formation and the efficiency of the reaction relies on multiple factors including the choice of protective groups and anomeric leaving groups (Fig. 1A).3 Thus, carbohydrate synthesis requires sophisticated chemical procedures and a great deal of technical expertise.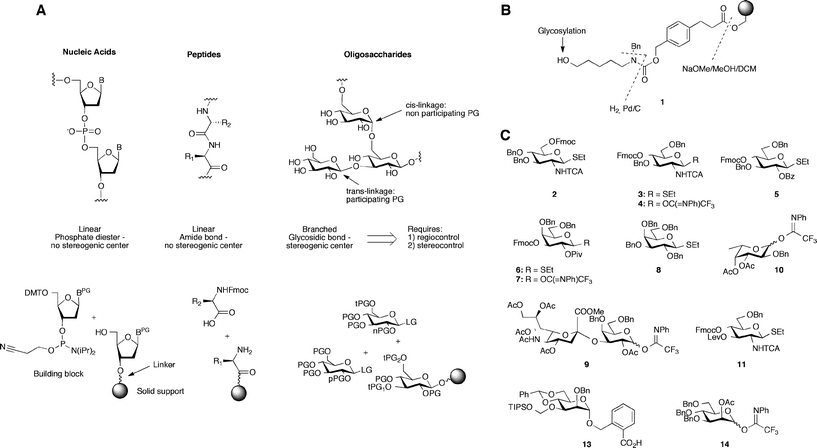 | ||
| Fig. 1 Fundamental considerations guiding automated solid phase oligosaccharide synthesis. (A) Solid-support synthesis of nucleic acids, peptides and oligosaccharides. Protecting groups: PG = protecting group; LG = leaving group; tPG = temporary protecting group; nPG = non-participating protecting group; pPG = participating protecting group; (B) linker design: new linker (structure 1) includes a C5-spacer and a latent terminal amine group revealed at the end of the synthesis by hydrogenolysis; (C) building blocks used for the construction of complex carbohydrates. | ||
Simple, efficient and reliable production of structurally-defined oligosaccharides based on a standardized, automated synthesis procedure will provide an essential foundation for advancing the field of glycobiology. To be of use in glycobiology studies, these oligosaccharides are usually immobilized or conjugated through a linker.4,5 Standardized oligosaccharide synthesis procedures, such as one-pot solution-phase assembly6–8 have been explored, but attempts towards automation have had limited success.9 Automated solution-phase assembly generated molecules up to hexasaccharides but serious engineering challenges remained.10,11 In 2001, we demonstrated the first proof-of-principle synthesizer for automated solid-phase oligosaccharide synthesis.12 Using a modified peptide synthesizer, and an octenediol linker to tether the nascent oligosaccharide chain to a polystyrene resin, differentially protected mono- and disaccharide building blocks were assembled into oligosaccharides.
This initial demonstration served to prove that automated assembly of increasingly complex oligosaccharides was possible. However, it also highlighted technical problems requiring innovative solutions founded in chemistry and engineering, to enable straightforward and user-friendly access to synthetic oligosaccharides. In particular, although large oligosaccharides were obtained, several complex post-assembly transformations would have been required to yield deprotected conjugation-ready structures for biological studies. Furthermore, severe shortcomings inherent to the use of an adapted peptide synthesizer precluded fully-automated synthesis. Here, we describe both a new synthesis strategy that limits solution phase manipulation as well as the first fully-automated solid-phase oligosaccharide synthesizer. Taken together, these constitute a versatile platform capable of generating complex conjugation-ready glycans (Fig. 2).
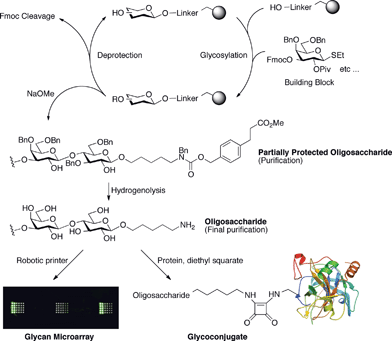 | ||
| Fig. 2 Overview of the fully-automated solid-phase oligosaccharide synthesis work flow. Building blocks are incorporated on the growing solid-support-bound structure in an iterative fashion. The assembled oligosaccharide is cleaved from the support in a form that is easily deprotected following a purification step. The final structure displays a spacer functionalized with a reactive amine that can be used to conjugate to proteins or microarrays. | ||
Results
Essential to the success of this platform was the creation of a fully-automated synthesis instrument that could support the synthesis strategy. This system combines solenoid valves for rapid pressure-driven washing steps with precisely-controlled syringe pump-driven reagent delivery. The machine was carefully designed so as to avoid mixing of incompatible reagents during liquid handling and to facilitate washing of the different lines. All vessels on the instrument are kept under an inert gas atmosphere to enable handling of reactive chemicals. The reaction vessel is a jacketed glass reactor connected to a cryostat. The temperature of the reaction vessel can be adjusted to as low as −50 °C and as high as 90 °C. Within the vessel, the solid support sits on a fritted glass filter. This setup allows for mixing via an inert gas stream introduced from the bottom of the filter as well as pressure-driven rapid removal of reaction mixtures and washing solvents. The outlet of the reaction vessel can be directed to waste or to a fraction collector. Importantly, this allows for the final cleavage product to be collected automatically. In addition, reaction mixtures can be collected and analyzed. Fully automated glycosylation and deprotection protocols (synthesis cycles) were designed whereby a computer program instructs the automated synthesizer to effect the glycosylation protocols for each building block, the key steps of which are reagent addition, solvent washes, and temperature regulation of the reaction vessel. In contrast, the previously used peptide synthesizer would require manual temperature adjustment at every glycosylation cycle and manual collection of reaction mixtures or cleaved product. Furthermore, the number of vessels for reagents was limited, therefore restricting the chemistry possible in a given synthesis. A detailed description of the synthesizer is provided in the ESI (Fig. S1–S4 and Schemes S1–S3).†Equally important was a synthesis strategy that integrated key elements such as the linker, building blocks, assembly and post-assembly manipulations. Central to this strategy is to release, upon assembly, a partially-protected oligosaccharide that can easily be purified and deprotected in a single hydrogenolysis step. The product of the final deprotection bears a primary amino group that is ready for conjugation to various supports for glycobiology studies. Solid-support oligosaccharide synthesis requires careful choice regarding the chemical composition of the linker that will connect the first sugar to the solid-support. The linker must not participate in unwanted reactions during glycosylations. A bi-functional linker 1 (Fig. 1B) was developed that was broadly chemically resilient, as its design was guided by the chemical parameters imposed by glycosylation reactions.13 Consequently, the linker is compatible with the most common glycosylating agents, including thioglycosides and glycosyl acetimidates. Cleavage of temporary hydroxyl protection groups such as 9-fluorenylmethoxycarbonyl (Fmoc), levulinoyl ester (Lev), and silyl ethers, as well as N-trichloroacetate protection of amine groups does not compromise linker integrity. An ester linkage attaches the linker to the solid-phase resin and is severed only after completion of synthesis by hydrolysis, which also cleaves all ester protective groups. This action also reveals the bifunctional architecture of the linker in the form of a latent C5-spacer with a unique terminal amine group, which will be used to attach the resultant oligosaccharides to microarrays, proteins or nanoparticles after complete deprotection (Fig. 2). After hydrolysis, the oligosaccharide is released bearing only protecting groups easily cleavable by hydrogenolysis.
Creation of the linker established the chemical boundaries for the construction of a collection of stable, differentially-protected glycan building blocks. Based on prevalence,14,15 an initial set of key building blocks were selected and synthesized on a large scale (Fig. 1C). The regiochemical and stereochemical information required to form the correct glycosidic bond, is contained within the monosaccharide building blocks. At loci bearing participating groups to direct the formation of trans-glycosidic linkages, benzoyl, acetyl, pivaloyl or N-trichloroacetyl groups are placed as appropriate. The versatility of our platform allowed us to exploit a wide variety of well-established glycosylation methods. Thioglycosides,16 carboxybenzyl glycosides,17 glycosyl trichloroacetimidates,18 and glycosyl N-phenyl trifluoroacetimidates19 were synthesized by drawing from previously established protocols (Fig. 1C, Schemes S5–S10†). In addition to the traditional, often time-consuming syntheses used for preparing most building blocks, a recently-introduced one-pot method for the streamlined synthesis of differentially protected thioglucoside building blocks was successfully adapted for these purposes to prepare novel thioglycosides on a multi-gram scale.20
For the assembly of resin-bound oligosaccharides, we built on protocols developed in our laboratory12,21–24 and developed new protocols for the use of thioglycosides. All building blocks are installed via an iterative, stepwise glycosylation/deprotection sequence (Fig. 2). Syntheses commence with glycosylation of the support-bound bifunctional linker with the first monosaccharide building block. Standardized glycosylation protocols were developed for each building block and were then used for each synthesis without further optimization. Glycosylation reactions are driven to completion using excess glycosylating agent and activator at temperatures that are dependent on the building block used. Glycoside bond formation is followed by washing steps. After this, at each elongation step, one (or more) protective group(s) is cleaved from the support-bound oligosaccharide to reveal the nucleophile(s) that will participate in the next glycosylation reaction. Capping and tagging25 steps were not used here, since for oligosaccharides of this size, separation of deletion sequences is not problematic. In line with our previous experience, Fmoc was chosen as a temporary protecting group for hydroxyl groups that will serve as nucleophiles during subsequent glycosylation steps and is readily cleaved by the action of piperidine in DMF.21 Levulinoyl esters (Lev), silyl ethers, and benzylidene acetals serve as temporary protective groups to allow for glycan chain branching.21,26,27
Post assembly, N-acetyl groups are revealed by reduction of the N-trichloroacetyl groups and the resin-bound oligosaccharide is cleaved and partially deprotected by hydrolysis treatment. These steps are performed by the synthesizer and result in oligomers that are, upon completion of the hydrolysis, protected only by benzyl ethers and benzyl amino carbamate. The benzyl groups provide a chromophoric and a hydrophobic handle for reverse-phase HPLC purification. Since the purification of partially-protected oligosaccharides by HPLC is still in its infancy compared to that of synthetic peptides and oligonucleotides, standard conditions for analytical and preparative reverse-phase HPLC were established. HPLC also served in determination of the product purity in particular since some oligomeric compounds give broad signals in NMR spectra. Finally, the permanent protective groups are removed by a simple hydrogenolysis at the last step of the synthesis. Purification by gel filtration or reverse-phase chromatography yields pure structurally-diverse oligosaccharides primed for simple conjugation to proteins or glycan microarray surfaces. A detailed description of the automated synthesis procedure can be found in the ESI.†
To illustrate the power and versatility of this new platform, diverse oligosaccharide targets were selected based on the following criteria: occurrence in glycospace, synthetic challenge, and biological relevance (Fig. 3).
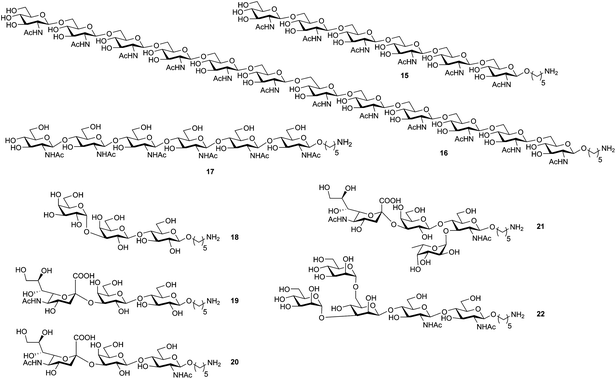 | ||
| Fig. 3 Oligosaccharide structures prepared by automated synthesis. 15 = β-(1→6)-glucosamine hexamer; 16 = dodecaglucosamine; 17 = β-(1→4)-glucosamine hexamer; 18 = iso-Gb3 trisaccharide; 19 = sialyl lactose; 20 = sialyl lactosamine; 21 = sialyl Lewisx; 22 = N-glycan core pentasaccharide. | ||
Oligo β-(1→6) and β-(1→4)-glucosamines (Fig. 3, compounds 15–17) are found on the surfaces of pathogenic bacteria that are part of biofilms.28 The bacterial β-(1→6)-glucosamine hexamer 15 (Fig. 3) was assembled using 6-Fmoc protected glucosamine thioglycoside building block 2 (Scheme S12†). Each cycle involved two additions of glycosylating agent (double coupling), using five equivalents of building block 2 per coupling in combination with the mild and non-toxic activator N-iodosuccinimide, in the presence of catalytic amounts of triflic acid at −20 °C.16 The temporary Fmoc protecting group was cleaved by the action of piperidine. Following assembly of the oligosaccharide chain, the C2 N-trichloroacetyl protective groups were reduced to the corresponding N-acetyl groups by six-fold exposure to tin hydride in the presence of the radical initiator azobisisobutyronitrile (AIBN). Release of the oligosaccharide product was achieved by cleavage of the linker with sodium methoxide. Preparative HPLC purification yielded 40 mg of the partially-protected hexasaccharide (Fig. S10†) corresponding to a 58% overall yield based on resin loading. Hydrogenolysis, as a single chemical transformation carried out manually, yielded 58% of fully deprotected hexasaccharide 15. Purification at the final stage was achieved by size exclusion chromatography. With step-wise coupling yields calculated at 96%, the synthesis of longer oligosaccharides was likely feasible. Dodecaglucosamine 16 (Fig. 3) therefore served as the next synthetic challenge. Employing the same automated protocol, the partially-protected dodecasaccharide was obtained with an overall yield of 43% before hydrogenolysis yielded dodecasaccharide 16 (Fig. S13†).
For the synthesis of β-(1→4)-glucosamine hexamer 17 (Fig. 3), two building blocks, glucosamine thioglycoside 3 and glycosyl N-phenyl trifluoroacetimidate 4, were compared for their efficiency in automated synthesis (Scheme S11†). While glucosamine N-phenyl trifluoroacetimidate 4 yielded the desired hexasaccharide as the main product, other oligosaccharide side-products were also obtained (Fig. S7†). Glucosamine thioglycoside 3 was prepared efficiently via a one-pot method20 and performed significantly better during automated oligosaccharide synthesis. The assembly yielded just small amounts of pentasaccharide deletion sequences and minor by-products in addition to the target hexasaccharide (Fig. S8†). Consequently, thioglycoside 3 with its associated glycosylation protocol was used for all subsequent syntheses containing this linkage. Preparative reverse-phase HPLC yielded 31% of the partially-protected oligosaccharide product overall. Hydrogenolysis followed by gel filtration furnished hexasaccharide 17 (Fig. S9†).
So far, all oligosaccharides that were assembled were the product of trans-glycosylations, which rely on participating protecting groups. The glycosphingolipid iso-Gb3 is involved in regulating the response of NKT cells to infection and malignancy29 and its trisaccharide portion (18, Fig. 3) served as an attractive target molecule to demonstrate that cis-glycosidic linkages can be installed. These linkage constructions rely on the anomeric effect in combination with the use of α-directing solvents.22 Using the thioglycoside building blocks 5, 6 and 8, the partially-protected trisaccharide was obtained. Installation of the α(1→3) galactoside was almost completely stereoselective as judged from the crude HPLC chromatogram (Fig. S14†). Preparative reverse-phase HPLC purification yielded 80% of the anomerically pure compound (Scheme S13†). Global deprotection by hydrogenolysis provided the desired trisaccharide 18 ready for conjugation.
Many naturally-occurring N- and O-glycans contain a terminal sialic acid that represents a synthetic challenge, as glycosylations involving sialic acid building blocks are commonly low yielding. Synthetic chemists often enlist help from enzymes to overcome this problem.30,31 A truly versatile oligosaccharide synthesis platform would ideally only utilize chemical steps to install the sialic acid caps. At this time however, no general chemical method exists to install a sialic acid reliably and with high efficiency on solid-support. With a preformed disaccharide as the only viable solution, we developed a sialic acid α-(2→3) galactose disaccharide building block 932 that can be efficiently installed as the terminal sialic acid residue. Two sialic acid-containing trisaccharides, sialyl lactose 19, and sialyl lactosamine 20, which are receptors for viral infection,33 were prepared to demonstrate the viability of this approach (Fig. 3, Schemes S14 and S15†). Compound 19 was produced with an overall yield of 36% and compound 20 with an overall yield of 26%.
The branching nature of oligosaccharides is a major distinguishing feature when compared to strictly linear oligopeptides and oligonucleotides (Fig. 1A). Sialyl Lewisx21 (Fig. 3), which is involved in inflammatory response,34 served to illustrate the synthesis of a branching structure and furthermore demonstrated that two types of building blocks, thioglycosides and glycosyl N-phenyl trifluoroacetimidates, can be employed in concert on the oligosaccharide synthesizer. Branching glucosamine building block 11 was prepared with C3-levulinate ester and C4-Fmoc carbonate protecting groups.21 Installation of glucosamine was followed by the addition of piperidine to cleave the Fmoc protecting group, unveiling the C4 position (Scheme S16†). Glycosylation using building block 9 was followed by removal of the levulinate ester to expose the second nucleophilic site on the central glucosamine. Glycosylation with 10 resulted in the partially-protected tetrasaccharide SI-27 (Scheme S16†) with an overall yield of 51% following hydrolysis and HPLC purification. Hydrogenolysis, was sufficient to remove all protective groups and produce tetrasaccharide 21.
The core pentasaccharide 22 (Fig. 4) common to all N-glycans has challenged many novel methods for oligosaccharide synthesis.35–40 With the notoriously difficult β(1→4)-mannosidic linkage connecting to the chitobiose disaccharide, and the two branches on the central mannose, this oligosaccharide was selected to showcase a combination of three different glycosylation methods in one synthesis, and thus challenge the platform. Previously, solid-phase syntheses of this particular structure had to resort to the use of disaccharides or other means to improve yields and selectivities.24 For the fully-automated synthesis, the reliable glucosamine thioglycoside 3 (Fig. 1C) was used to install the chitobiose portion. The crucial β-mannosidic linkage was introduced by coupling the carbyloxy benzyl glycoside building block 13 (Fig. 1C) carrying both a tert-butyldimethylsilyloxymethyl (tom) ether at C3, as well as a benzylidene acetal protecting group to mask the C4 and C6 hydroxyl groups.23 After removal of the tom group on C3 by the action of fluoride, the benzylidene acetal was selectively opened to reveal the C6 hydroxyl group using borane. Bis-mannosylation was achieved using mannose N-phenyl trifluoroacetimidate building block 14 (Fig. 1C) that requires activation with trimethylsilyl triflate at 10 °C to finish pentasaccharide assembly (Fig. 4). Reverse-phase HPLC-MS analysis of the partially-protected oligosaccharide 23 following cleavage from the resin revealed pentasaccharide as the major product, albeit as a mixture of α- and β-anomers that resulted from the modestly selective β-mannosylation reaction (Fig. 4, inserts a and b). The poor yield of the overall synthesis (3.5%) can be attributed to a combination of low efficiency in the non-standard steps necessary for this synthesis, as well as the extensive manipulations that were required to identify appropriate purification methods. Using preparative HPLC, the pentasaccharide anomers were cleanly separated (Fig. S21†) to give the desired β-mannoside-containing pentasaccharide. Routine global deprotection by hydrogenolysis yielded 22 in 78% yield.
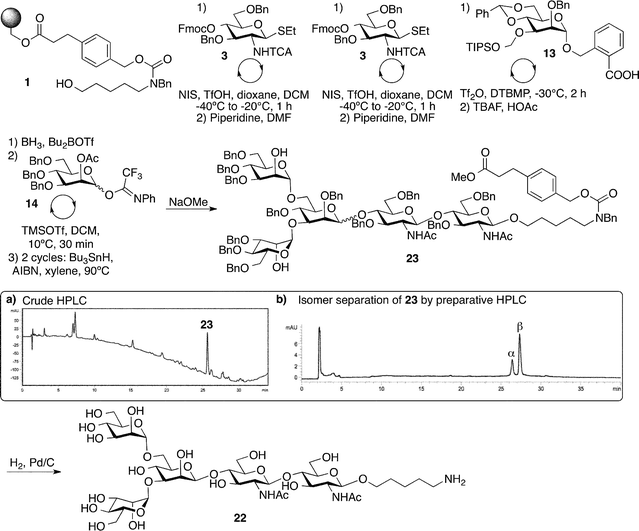 | ||
| Fig. 4 Automated synthesis of the N-glycan core structure 22. Insert (a) shows the HPLC chromatogram of the crude material. Insert (b) shows the HPLC chromatogram of the mixture of α- and β-anomers separated on a YMC-pack diol column. | ||
Selected glycans provided by the automated platform were conjugated to a microarray and a protein to illustrate their utility in the production of tools for glycobiology. The glycan microarray was printed using a piezoelectric robotic printer. Subsequent incubation with the lectin wheat germ agglutinin (WGA) confirmed the conjugation (Fig. S23†).5 Oligosaccharide 17 was conjugated to bovine serum albumin (BSA) using the squarate ester method (Fig. S22†).41 Such glycan–protein conjugates are important for the production of anti-glycan antibodies and can serve as constituents of conjugate vaccines.4,42
Conclusions
In summary, we have developed a platform integrating a novel synthesis strategy and a new fully-automated oligosaccharide synthesizer. Central to the strategy is the release of a partially-protected glycan that upon purification can be deprotected in a single step to yield a conjugation-ready glycan. The versatile platform we described here will serve as a basis to improve specific facets of the automated assembly process, in order to ultimately provide automated access to even more challenging classes of carbohydrates, including glycosaminoglycans and bacterial oligosaccharides.Acknowledgements
Generous financial support from the Max-Planck Society and the Swiss National Science Foundation (200020-117889) as well as a Körber Prize (to P. H. Seeberger), and fellowships from the Roche Research Foundation (to L. Kröck), the National Science Council Taiwan (to C.-C. Wang), and Novartis (to P. Bindschädler) are gratefully acknowledged. We thank Dr W. C. Christ (Ancora Pharmaceuticals) for useful discussions regarding the design of the reaction vessel. We thank Dr V. Mountain for critically editing the manuscript.Notes and references
- R. B. Merrifield, Science, 1965, 150, 178–185 CAS.
- M. H. Caruthers, Science, 1985, 230, 281–285 CAS.
- X. Zhu and R. R. Schmidt, Angew. Chem., Int. Ed., 2009, 48, 1900–1934 CrossRef CAS.
- G. J. Bernardes, B. Castagner and P. H. Seeberger, ACS Chem. Biol., 2009, 4, 703–713 CrossRef CAS.
- J. Stevens, O. Blixt, J. C. Paulson and I. A. Wilson, Nat. Rev. Microbiol., 2006, 4, 857–864 CrossRef CAS.
- Y. Wang, X. S. Ye and L. H. Zhang, Org. Biomol. Chem., 2007, 5, 2189–2200 CAS.
- J. C. Lee, W. A. Greenberg and C. H. Wong, Nat. Protoc., 2007, 1, 3143–3152 CrossRef.
- T. K. Mong, H. K. Lee, S. G. Duron and C. H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 797–802 CrossRef CAS.
- P. H. Seeberger and W. C. Haase, Chem. Rev., 2000, 100, 4349–4394 CrossRef CAS.
- S. P. Douglas, D. M. Whitfield and J. J. Krepinsky, J. Am. Chem. Soc., 1991, 113, 5095–5097 CrossRef CAS.
- F. A. Jaipuri and N. L. Pohl, Org. Biomol. Chem., 2008, 6, 2686–2691 CAS.
- O. J. Plante, E. R. Palmacci and P. H. Seeberger, Science, 2001, 291, 1523–1527 CrossRef CAS.
- P. Czechura, N. Guedes, S. Kopitzki, N. Vazquez, M. Martin-Lomas and N.-C. Reichardt, Chem. Commun., 2011, 47, 2390 RSC.
- D. B. Werz, R. Ranzinger, S. Herget, A. Adibekian, C. W. von der Lieth and P. H. Seeberger, ACS Chem. Biol., 2007, 2, 685–691 CrossRef CAS.
- A. Adibekian, P. Stallforth, M.-L. Hecht, D. B. Werz, P. Gagneux and P. H. Seeberger, Chem. Sci., 2011, 2, 337–344 RSC.
- J. D. Codee, R. E. Litjens, L. J. van den Bos, H. S. Overkleeft and G. A. van der Marel, Chem. Soc. Rev., 2005, 34, 769–782 RSC.
- K. S. Kim, J. H. Kim, Y. J. Lee and J. Park, J. Am. Chem. Soc., 2001, 123, 8477–8481 CrossRef CAS.
- R. R. Schmidt and J. Michel, Angew. Chem., Int. Ed. Engl., 1980, 19, 731–732 CrossRef.
- B. Yu and H. C. Tao, Tetrahedron Lett., 2001, 42, 2405–2407 CrossRef CAS.
- C. C. Wang, J. C. Lee, S. Y. Luo, S. S. Kulkarni, Y. W. Huang, C. C. Lee, K. L. Chang and S. C. Hung, Nature, 2007, 446, 896–899 CrossRef CAS.
- K. Routenberg Love and P. H. Seeberger, Angew. Chem., Int. Ed., 2004, 43, 602–605 CrossRef.
- D. B. Werz, B. Castagner and P. H. Seeberger, J. Am. Chem. Soc., 2007, 129, 2770–2771 CrossRef CAS.
- J. D. C. Codee, L. Krock, B. Castagner and P. H. Seeberger, Chem.–Eur. J., 2008, 14, 3987–3994 CrossRef CAS.
- D. M. Ratner, E. R. Swanson and P. H. Seeberger, Org. Lett., 2003, 5, 4717–4720 CrossRef CAS.
- F. R. Carrel and P. H. Seeberger, J. Org. Chem., 2008, 73, 2058–2065 CrossRef CAS.
- S. D. Markad and R. R. Schmidt, Eur. J. Org. Chem., 2009, 5002–5011 CrossRef CAS.
- T. Zhu and G.-J. Boons, Chem.–Eur. J., 2001, 7, 2382–2389 CrossRef CAS.
- T. Maira-Litran, A. Kropec, D. Goldmann and G. B. Pier, Vaccine, 2004, 22, 872–879 CrossRef CAS.
- D. Zhou, J. Mattner, C. Cantu, 3rd, N. Schrantz, N. Yin, Y. Gao, Y. Sagiv, K. Hudspeth, Y. P. Wu, T. Yamashita, S. Teneberg, D. Wang, R. L. Proia, S. B. Levery, P. B. Savage, L. Teyton and A. Bendelac, Science, 2004, 306, 1786–1789 CrossRef CAS.
- B. Ernst and R. Oehrlein, Glycoconjugate J., 1999, 16, 161–170 CrossRef CAS.
- H. A. Chokhawala, S. Huang, K. Lau, H. Yu, J. Cheng, V. Thon, N. Hurtado-Ziola, J. A. Guerrero, A. Varki and X. Chen, ACS Chem. Biol., 2008, 3, 567–576 CrossRef CAS.
- S. Hanashima, B. Castagner, D. Esposito, T. Nokami and P. H. Seeberger, Org. Lett., 2007, 9, 1777–1779 CrossRef CAS.
- S. Hanashima and P. H. Seeberger, Chem.–Asian J., 2007, 2, 1447–1459 CrossRef CAS.
- R. Kannagi, M. Izawa, T. Koike, K. Miyazaki and N. Kimura, Cancer Sci., 2004, 95, 377–384 CrossRef CAS.
- H. Paulsen, Angew. Chem., Int. Ed. Engl., 1990, 29, 823–839 CrossRef.
- S. J. Danishefsky, S. Hu, P. F. Cirillo, M. Eckhardt and P. H. Seeberger, Chem.–Eur. J., 1997, 3, 1617–1628 CrossRef CAS.
- S. Eller, R. Schuberth, G. Gundel, J. Seifert and C. Unverzagt, Angew. Chem., Int. Ed., 2007, 46, 4173–4175 CrossRef CAS.
- I. Matsuo, M. Wada, S. Manabe, Y. Yamaguchi, K. Otake, K. Kato and Y. Ito, J. Am. Chem. Soc., 2003, 125, 3402–3403 CrossRef CAS.
- X. Y. Wu, M. Grathwohl and R. R. Schmidt, Angew. Chem., Int. Ed., 2002, 41, 4489–4493 CrossRef CAS.
- S. Jonke, K. G. Liu and R. R. Schmidt, Chem.–Eur. J., 2006, 12, 1274–1290 CrossRef CAS.
- V. P. Kamath, P. Diedrich and O. Hindsgaul, Glycoconjugate J., 1996, 13, 315–319 CrossRef CAS.
- R. Roy, Drug Discovery Today: Technol., 2004, 1, 327–336 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures: a complete description of the automated oligosaccharide synthesizer set-up and operations. Also described are the linker synthesis, building block syntheses, automated syntheses and preparation of glycoconjugates. See DOI: 10.1039/c2sc00940d. |
| ‡ Present address: Institute of Pharmaceutical Sciences, Swiss Federal Institute of Technology (ETH) Zurich, 8093 Zurich, Switzerland. |
| § Present address: Institute of Chemistry, Academia Sinica, Taipei, 11529, Taiwan. |
| ¶ Present address: BASF SE, GVA/IO–B009, Carl-Bosch-Str. 38, 67056 Ludwigshafen, Germany. |
| This journal is © The Royal Society of Chemistry 2012 |