DOI:
10.1039/C1SC00458A
(Edge Article)
Chem. Sci., 2012,
3, 244-248
Received
12th July 2011
, Accepted 8th September 2011
First published on 23rd September 2011
Abstract
A series of quinoline and tetrahydroquinoline derivatives were prepared via a one-pot protocol involving intramolecular palladium(0)-catalyzed cyclopropane sp3 C–H bond functionalization and subsequent oxidation or reduction. The reaction conditions demonstrate good tolerance for a wide range of functional groups. Evidence suggests that the reaction proceeds through C–H bond cleavage and C–C bond formation to generate a dihydroquinoline intermediate via in situcyclopropane ring-opening.
Introduction
Over the past decade, the palladium(0)-catalyzed transformation of C–H bonds into C–C bonds has attracted considerable attention due to its ability to streamline organic synthesis.1 Such processes avoid the need for stoichiometric organometallic reagents as coupling partners along with any problems associated with their synthesis, stability, and/or functional group compatibility.2
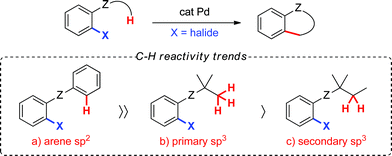
While both sp2 and sp3 C–H bonds can be functionalized, the latter are less reactive since they do not benefit from favourable catalyst–substrate interactions via an arene π-electron cloud.3 In the context of palladium(0)-catalyzed arylation at sp3 C–H bonds, reports have overwhelmingly centered on the functionalization of methyl groups, while methylene arylation has been reported only on very rare occasions (Scheme 1).4 Although methylene functionalization has been described using Pd(II)-catalysis,5 the tolerance for a wide range of functional groups in Pd(0)-catalysis and the ability to use inexpensive (or easily accessible) aryl bromides or chlorides as coupling partners present an attractive complementary alternative.
 |
| | Scheme 1
Palladium(0)-catalyzed intramolecular C–C bond forming reactions involving C–H bonds of varying hybridization. | |
Towards this end, we chose to investigate the reactivity of secondary cyclopropane C–H bonds, which may provide opportunities and insight towards more general palladium(0)-catalyzed methylene C–H arylations.6–8 The inherent ring strain and orbital rehybridization found within cyclopropane ring systems9 may facilitate the desired transformation. For example, the carbon atoms in cyclopropane rings exhibit sp2-like properties, which may provide the advantage of greater catalyst–substrate interactions and, therefore, promote C–H bond cleavage.
Herein, we describe the palladium(0)-catalyzed intramolecular arylation of cyclopropane methylene C–H bonds to generate 1,4-dihydroquinolines via in situ ring-opening (Scheme 2). The reaction mixture can subsequently be treated under oxidative or reductive conditions to afford the corresponding quinolines or 1,2,3,4-tetrahydroquinolines, prevalent motifs in natural products, medicinal chemistry and material science.10 Moreover, despite the significant diversity of regioselective methods for their preparation,11,12 those that provide C2, C3 and C4 substituent-free derivatives are rare.13–15
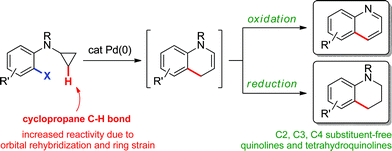 |
| | Scheme 2 Preparation of quinolines and tetrahydroquinolines via an sp3 C–H bond functionalization strategy. | |
Results and discussion
Synthesis of quinolines and tetrahydroquinolines
On the basis of our previous work on intramolecular alkane arylation of primary C–H bonds (Scheme 1b),3b,g,h initial studies focused on the optimization of reaction conditions for the palladium-catalyzed C–H arylation of bromophenyl cyclopropyl carbamate 1a (Table 1).16 Upon examination of this process, the base and pivalate additive were found to have a profound effect on the reaction yield.17 Optimal conditions for the formation of 1,4-dihydroquinoline 2a employed a combination of palladium acetate (5 mol%), di-tert-butyl(methyl)phosphine (as its tetrafluoroborate salt) (10 mol%), caesium pivalate (30 mol%) and potassium phosphate (1.5 equivalents) in mesitylene at 90 °C for 16 h (Conditions A). After isolation, product 2a rapidly decomposed,18 leading us to investigate the viability of a subsequent same-pot oxidation protocol to quinoline 3a (Table 1). Upon further optimization, it was found that the cooled reaction mixture could be diluted with THF and treated with DDQ (1.2 equivalents) to afford the aromatized/oxidized product 3a in 87% isolated yield over two steps (entry 1).
|
Conditions A:
Pd(OAc)2 (5 mol%), P(tBu)2Me·HBF4 (10 mol%), CsOPiv (30 mol%), K3PO4 (1.5 equiv.), 90–110 °C, 16 h. Conditions B:Pd(OAc)2 (5 mol%), PCy3·HBF4 (10 mol%), PivOH (30 mol%), Cs2CO3 (1.5 equiv.), 110–140 °C, 3–16 h.
DDQ (1.2 equiv.), THF, 0 °C to rt, 2–20 h.
Isolated yield.
N-protecting group “CO2Me” was replaced by “Cbz”.
|

|
These conditions for C–H arylation and subsequent oxidation, with minor variations in temperature (90–110 °C), were applied to aryl bromides 1b–h bearing electron-donating or -withdrawing groups at the ortho, meta or para positions to provide the corresponding C5, C6 or C7-substituted quinolines 3b–h in moderate to very good yields (entries 2–9). Steric hindrance adjacent to the bromide was well tolerated as exemplified by the formation of 3h in 87% yield (entry 9). The methylcarbamate protecting group could be replaced by a benzylcarbamate (Cbz) albeit at a minor cost in yield (entries 2 and 3).
Aryl chlorides 1i–l were also found to be suitable coupling partners for this transformation, after slightly modifying the conditions for the palladium-catalyzed coupling reaction (Conditions B). The use of tricyclohexylphosphine as the ligand, pivalic acid as the additive and caesium carbonate as the base, with an increase in reaction temperature (110–140 °C), allowed for the preparation of other diversely substituted quinolines 3a,j–l in good yields (entries 10–13). Of note is the tolerance of this method for functional groups that may not be compatible with some of the more traditional (harsh) acid-catalyzed quinoline syntheses (entries 11 and 13).13
The formation of 1,4-dihydroquinoline intermediates 2 also provides an opportunity for the preparation of 1,2,3,4-tetrahydroquinoline analogues (Table 2). For example, N-protected tetrahydroquinolines 4a,d,h,l were readily obtained from the corresponding aryl bromides (entries 1–3) or chloride (entry 4) viaPd(0)-catalyzed C–C bond formation, followed by a simple reductive treatment of dihydroquinoline intermediate 2 (using catalytic Pd/C under an atmosphere of H2) in a one-pot fashion.
|
Conditions A:
Pd(OAc)2 (5 mol%), P(tBu)2Me·HBF4 (10 mol%), CsOPiv (30 mol%), K3PO4 (1.5 equiv.), 90–110 °C, 16 h; Conditions B:Pd(OAc)2 (5 mol%), PCy3·HBF4 (10 mol%), PivOH (30 mol%), Cs2CO3 (1.5 equiv.), 140 °C, 3 h.
Pd/C (10 mol%), H2 (balloon pressure), EtOAc, rt, 12–24 h.
Isolated yield.
Intermediate dihydroquinoline 2d was also isolated in 53% yield.
|

|
Moreover, the scope of the Pd(0)-catalyzed arylation of secondary C–H bonds was further examined with respect to the heteroatom in the tether between the arene and the cyclopropyl moieties. Using Conditions B at 110 °C, an oxygen atom in the tether was well tolerated, yielding benzochromene 6 in 91% yield (eqn (1)).
| |  | (1) |
Mechanistic discussion
Two major pathways emerged as potential explanations for the formation of dihydroquinoline 2a from cyclopropylaniline 1a (Scheme 3). Following oxidative addition, intermediate A could undergo cyclopropane ring-opening promoted by an electrophilic Pd(II) center (Path a).19 The resulting palladacycle B could then be deprotonated, and subsequent reductive elimination would provide dihydroquinoline 2a. Alternatively, oxidative addition could be followed by a ligand exchange, generating pivalate-bound Pd(II) species A′. The latter can lead to 2aviapivalate-promoted sp3 C–H bond cleavage,20 a process that has been previously demonstrated for the arylation of primary sp3 C–H bonds (Path b).3b,g Six-membered ring palladacycle C could then reductively eliminate product 7a, which may ring-open under the reaction conditions to generate the corresponding dihydroquinoline. However, reductive elimination to form the highly strained ring-system in 7a may be challenging. Thus, cyclopropane ring-opening to generate Bvia intramolecular proton transfer, with concomitant release of ring strain, may be favoured.21
Based on these hypotheses, experiments were conducted to distinguish between the possible pathways for this transformation (eqn (2)–(6)). When a standard reaction involving cyclopropylaniline 1a was run in the absence of a pivalate additive, no dihydroquinoline product 2a could be detected (eqn (2)). The use of carboxylate additives in Pd(0)-catalyzed C–H bond functionalizations is well precedented and is consistent with a C–H bond-breaking event occurring via a concerted metallation–deprotonation (CMD) transition-state (Scheme 3, Path b).20 Furthermore, trace amounts of tetrahydrocyclopropa[b]indole 7a were isolated (eqn (3)), leading us to investigate its role, either as a potential intermediate or as a reaction by-product. Compound 7a, independently prepared by Simmons–Smith cyclopropanation of the corresponding N-protected indole, was submitted to the standard reaction conditions and proved to be very stable; cyclopropane ring-opening was not observed, even at temperatures as high as 140 °C (eqn (4)).22 These results suggest that dihydroquinoline formation occurs through a three-step sequence (Scheme 3): i) pivalate-promoted sp3 C–H bond cleavage, ii) cyclopropane C–C bond cleavage/ring-opening and, iii) deprotonation and reductive elimination.
| | 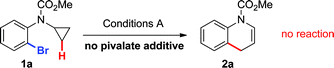 | (2) |
| | 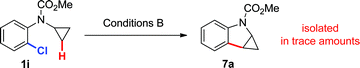 | (3) |
| |  | (4) |
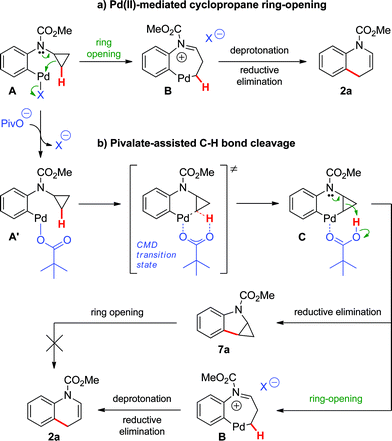 |
| | Scheme 3 Proposed mechanisms for the Pd(0)-catalyzed formation of dihydroquinoline 2a. | |
Finally, to provide additional evidence for the formation of 2avia Path b, the arylations of cyclobutane 8 and cyclopropane 10 were investigated. Both substrates may generate palladacycles analogous to C but with less ring strain.23 Therefore, we hypothesized that these intermediates may be less prone to ring-open prior to reductive elimination, providing direct evidence for methylene C–H bond functionalization. Under standard reactions conditions (Conditions B, 140 °C), indoline 9 was isolated in 65% yield (eqn (5)), while an inseparable mixture of dihydrobenzofuran 11 and chroman 12 was obtained in 67% yield (in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) (eqn (6)).
:1 ratio) (eqn (6)).
| | 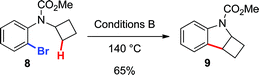 | (5) |
| | 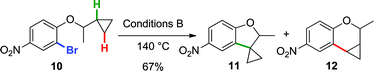 | (6) |
Conclusion
In summary, a one-pot quinoline and tetrahydroquinoline synthesis from cyclopropylanilines has been developed, employing a palladium(0)-catalyzed arylation of secondary aliphatic C–H bonds, and subsequent one-pot oxidation or reduction, respectively. Mechanistic experiments suggest that sp3 C–H bond functionalization occurs prior to cyclopropane ring-opening. Future studies will aim to broaden the scope of the Pd(0)-catalyzed arylation of secondary aliphatic C–H bonds, focusing on elucidating the factors that govern this reactivity.
Acknowledgements
We thank NSERC and the University of Ottawa for support of this work. Eli Lilly, Amgen and Astra Zeneca Montreal are thanked for unrestricted financial support. S.R. thanks NSERC for a CGS-D postgraduate fellowship. We also thank Prof. Stephen Buchwald and Prof. Louis Barriault for enabling the completion of this project. Dr David Stuart and Dr Brett Fors are acknowledged for insightful discussions and critical reading of this manuscript.
Notes and references
- Selected recent reviews on transition metal-catalyzed C–H bond functionalization:
(a) P. Thansandote and M. Lautens, Chem.–Eur. J., 2009, 15, 5874–5833 CrossRef CAS;
(b) R. Jazzar, J. Hitce, A. Renaudat, J. Sofack-Kreutzer and O. Baudoin, Chem.–Eur. J., 2010, 16, 2654–2672 CrossRef CAS;
(c) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS;
(d)
C–H Activation, ed. J.-Q. Yu and Z.-J. Shi, Top. Curr. Chem., 2010, 292 Search PubMed;
(e) H. Li, B.-J. Li and Z.-J. Shi, Catal. Sci. Technol., 2011, 1, 191–206 RSC;
(f) O. Baudoin, Chem. Soc. Rev. 10.1039/c1cs15058h.
- Selected reviews on transition metal-catalyzed cross-coupling reactions:
(a) J. Hassan, M. Sévignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359–1470 CrossRef CAS;
(b)
Metal-Catalyzed Cross-Coupling Reactions, ed. F. Diederich and P. J. Stang, Wiley-VCH, Weinheim, 2004 Search PubMed.
-
Pd(0)-catalyzed C–C bond forming reactions from non-acidic sp3 C–H bonds:
(a) J. Hitce, P. Retailleau and O. Baudoin, Chem.–Eur. J., 2007, 13, 792–799 CrossRef CAS;
(b) M. Lafrance, S. I. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2007, 129, 14570–14571 CrossRef CAS;
(c) T. Watanabe, S. Oishi, N. Fujii and H. Ohno, Org. Lett., 2008, 10, 1759–1762 CrossRef CAS;
(d) H. S. Kim, S. Gowrisankar, S. H. Kim and J. N. Kim, Tetrahedron Lett., 2008, 49, 3858–3861 CrossRef CAS;
(e) M. Chaumontet, R. Piccardi, N. Audic, J. Hitce, J.-L. Peglion, E. Clot and O. Baudoin, J. Am. Chem. Soc., 2008, 130, 15157–15166 CrossRef CAS;
(f) M. Wasa, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 9886–9887 CrossRef CAS;
(g) S. Rousseaux, S. I. Gorelsky, B. K. W. Chung and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 10692–10705 CrossRef CAS;
(h) S. Rousseaux, M. Davi, J. Sofack-Kreutzer, C. Pierre, C. E. Kefalidis, E. Clot, K. Fagnou and O. Baudoin, J. Am. Chem. Soc., 2010, 132, 10706–10716 CrossRef CAS;
(i) C. Pierre and O. Baudoin, Org. Lett., 2011, 13, 1816–1819 CrossRef CAS;
(j) M. Nakanishi, D. Katayev, C. Besnard and E. P. Kündig, Angew. Chem., Int. Ed., 2011, 50, 7438–7441 CrossRef CAS.
-
Pd(0)-catalyzed arylations of unactivated methylene C–H bonds have been reported on rare occasions; see ref. 3c, d, j.
-
Pd(II)-catalyzed arylation of unactivated methylene C–H bonds:
(a) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154–13155 CrossRef CAS;
(b) B. V. S. Reddy, L. R. Reddy and E. J. Corey, Org. Lett., 2006, 8, 3391–3394 CrossRef CAS;
(c) D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965–3972 CrossRef CAS;
(d) Y. Feng, Y. Wang, B. Landgraf, S. Liu and G. Chen, Org. Lett., 2010, 12, 3414–3417 CrossRef CAS;
(e) G. He and G. Chen, Angew. Chem., Int. Ed., 2011, 50, 5192–5196 CrossRef CAS.
- For examples of Pd(II)-catalyzed cyclopropane C–H bond functionalization, see:
(a) R. Giri, X. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2005, 44, 2112–2115 CrossRef CAS;
(b) R. Giri, N. Maugel, J.-J. Li, D.-H. Wang, S. P. Breazzano, L. B. Saunders and J.-Q. Yu, J. Am. Chem. Soc., 2007, 129, 3510–3511 CrossRef CAS;
(c) D.-H. Wang, M. Wasa, R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 7190–7191 CrossRef CAS;
(d) M. Wasa, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 3680–3681 CrossRef CAS;
(e) E. J. Yoo, M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 17378–17380 CrossRef CAS;
(f) K. J. Stowers, K. C. Fortner and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 6541–6544 CrossRef CAS;
(g) A. Kubota and M. S. Sanford, Synthesis, 2011, 2579–2589 Search PubMed.
- For an example of Ru-catalyzed carbonylation of a cyclopropane C–H bond, see: N. Hasegawa, V. Charra, S. Inoue, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2011, 133, 8070–7073 CrossRef CAS.
- M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117–3179 CrossRef CAS.
- K. Exner and P.v. R. Schleyer, J. Phys. Chem. A, 2001, 105, 3407–3416 CrossRef CAS , and references therein.
-
(a) A. Kimyonok, X.-Y. Wang and M. Weck, Polym. Rev., 2006, 46, 47–77 CAS;
(b) J. P. Michael, Nat. Prod. Rep., 2008, 25, 166–187 RSC;
(c) S. Kumar, S. Bawa and G. Himanshu, Mini-Rev. Med. Chem., 2009, 9, 1648–1654 CrossRef CAS;
(d) A. Danel, E. Gondek and I. Kityk, Opt. Mater., 2009, 32, 267–273 CrossRef CAS;
(e) K. Kaur, M. Jain, R. P. Reddy and R. Jain, Eur. J. Med. Chem., 2010, 45, 3245–3264 CrossRef CAS.
-
(a)
Name Reactions in Heterocyclic Chemistry, ed. J. J. Li, John Wiley & Sons, Hoboken, NJ, 2005, pp. 386–456 Search PubMed;
(b)
Heterocyclic Chemistry, 5th edn, ed. J. A. Joule and K. Mills, John Wiley & Sons, Chichester, UK, 2010, pp. 177–204 Search PubMed;
(c) V. Sridharan, P. A. Suryavanshi and J. C. Menéndez, Chem. Rev., 2011 DOI:10.1021/cr100307m.
- Selected recent transition metal-catalyzed quinoline syntheses:
(a) M. Movassaghi and M. D. Hill, J. Am. Chem. Soc., 2006, 128, 4592–4593 CrossRef CAS;
(b) X.-Y. Lui, P. Ding, J.-S. Huang and C.-M. Che, Org. Lett., 2007, 9, 2645–2648 CrossRef;
(c) Z. Zhang, J. Tan and Z. Wang, Org. Lett., 2008, 10, 173–175 CrossRef CAS;
(d) A. Arcadi, M. Aschi, F. Marinelli and M. Verdecchia, Tetrahedron, 2008, 64, 5354–5361 CrossRef CAS;
(e) H. Waldmann, G. V. Karunakar and K. Kumar, Org. Lett., 2008, 10, 2159–2162 CrossRef CAS;
(f) J. Horn, S. P. Marsden, A. Nelson, D. House and G. G. Weingarten, Org. Lett., 2008, 10, 4117–4120 CrossRef CAS;
(g) K. Cao, F.-M. Zhang, Y.-Q. Tu, X.-T. Zhuo and C.-A. Fan, Chem.–Eur. J., 2009, 15, 6332–6334 CrossRef CAS;
(h) H. Huang, H. Jiang, K. Chen and H. Lui, J. Org. Chem., 2009, 74, 5476–5480 CrossRef CAS;
(i) S. Majumder, K. R. Gipson and A. L. Odom, Org. Lett., 2009, 11, 4720–4723 CrossRef CAS;
(j) M. Zhang, T. Roisnel and P. H. Dixneuf, Adv. Synth. Catal., 2010, 352, 1896–1903 CrossRef CAS;
(k) R. N. Monrad and R. Madsen, Org. Biomol. Chem., 2011, 9, 610–615 RSC.
- Among the traditional methods,11a,b the Skraup synthesis can lead to quinolines bearing no substituents on the pyridine ring. However, this reaction typically suffers from moderate product yields and the use of harsh reaction conditions often limits the substrate scope and applications on large scale. See: D. A. Conlon, A. Drahus-Paone, G.-J. Ho, B. Pipik, R. Helmy, J. M. McNamara, Y.-J. Shi, J. M. Williams, D. Macdonald, D. Deschênes, M. Gallant, A. Mastracchio, B. Roy and J. Scheigetz, Org. Process Res. Dev., 2006, 10, 36–45 CrossRef CAS , and references therein.
- For other isolated examples of C2, C3 and C4 substituent-free quinoline syntheses, see:
(a) Y. Tsuji, H. Nishimura, K.-T. Huh and Y. Watanabe, J. Organomet. Chem., 1985, 286, c44–c46 CrossRef CAS;
(b) Y. Tsuji, K.-T. Huh and Y. Watanabe, J. Org. Chem., 1987, 52, 1673–1680 CrossRef CAS;
(c) I.-S. Cho, L. Gong and J. M. Muchowski, J. Org. Chem., 1991, 56, 7288–7291 CrossRef CAS;
(d) C. S. Cho, B. H. Oh and S. C. Shim, J. Heterocycl. Chem., 1999, 36, 1175–1178 CrossRef CAS;
(e) C. S. Cho, T. K. Kim, T.-J. Kim and S. C. Shim, J. Heterocycl. Chem., 2002, 39, 291–294 CrossRef CAS;
(f) F. Portela-Cubillo, J. S. Scott and J. C. Walton, J. Org. Chem., 2008, 73, 5558–5565 CrossRef CAS;
(g) H. Aramoto, Y. Obora and Y. Ishii, J. Org. Chem., 2009, 74, 628–633 CrossRef CAS.
- For examples of C2, C3 and C4 substituent-free tetrahydroquinoline syntheses, see ref. 11c and references therein.
- See the ESI† for the preparation of starting materials.
- The use of K2CO3 with CsOPiv or Cs2CO3 with PivOH were also viable options, albeit providing 2a in lower yields than the use of K3PO4 with CsOPiv.
- See the ESI† for the characterization of this intermediate. See also: T. Watanabe, S. Oishi, N. Fujii and H. Ohno, Org. Lett., 2007, 9, 4821–4824 CrossRef CAS.
- Selected recent examples of Pd-mediated or -catalyzed ring-opening reactions of substituted cyclopropanes:
(a) S.-B. Park and J. K. Cha, Org. Lett., 2000, 2, 147–149 CrossRef CAS;
(b) H. Okumoto, T. Jinnai, H. Shimizu, Y. Harada, H. Mishima and A. Suzuki, Synlett, 2000, 629–630 CAS;
(c) Z. He and A. K. Yudin, Org. Lett., 2006, 8, 5829–5832 CrossRef CAS;
(d) M. Shan and G. A. O'Doherty, Org. Lett., 2008, 10, 3381–3384 CrossRef CAS;
(e) D. Rosa and A. Orellana, Org. Lett., 2011, 13, 110–113 CrossRef CAS . See also ref. 6g.
- For reviews, see:
(a) Y. Boutadla, D. L. Davies, S. A. Macgregor and A. I. Poblador-Bahamonde, Dalton Trans., 2009, 5820–5831 RSC;
(b) D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118–1126 CrossRef;
(c) L. Ackermann, Chem. Rev., 2011, 111, 1315–1645 CrossRef CAS.
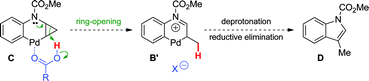
- Alternatively, ring-opening of intermediate C could lead to 6-membered palladacycle B′, ultimately providing 3-methylindole D.
However, this indole by-product has not been observed either by GC-MS or 1H NMR; ring-opening to B′via intramolecular proton transfer may be disfavoured due to stereochemical effects (i.e. the acidic proton and the cyclopropane methylene carbon in intermediate C lie on opposite sides of the plane formed by the rigid 6-membered palladacycle). We thank a reviewer for pointing out this mechanistic pathway.
- To eliminate the possibility of ring-opening by a catalytic species, 7a was also submitted to the standard reaction conditions in the presence of aryl chloride 1l. While quinoline 3l was produced in 60% yield (1H NMR), compound 7a remained intact.
- For a recent review about cyclobutanes in catalysis, see: T. Seiser, T. Saget, D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 7740–7752 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, characterization data, and NMR spectra for all new compounds. See DOI: 10.1039/c1sc00458a |
| ‡ Current address: Institut Charles Gerhardt, UMR-CNRS 5253, ENSCM, AM2N, 8 rue de l'école normale, 34296 Montpellier Cedex 5, France. |
| § Prof. Keith Fagnou passed away on November 11, 2009. |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.