Clickable, photoreactive inhibitors to probe the active site microenvironment of fatty acid amide hydrolase†
Susanna M.
Saario
,
Michele K.
McKinney
,
Anna E.
Speers
,
Chu
Wang
and
Benjamin F.
Cravatt
*
The Scripps Research Institute, 10550 N. Torrey Pines Road, La Jolla, CA 92037. E-mail: cravatt@scripps.edu; Fax: +858-784-8223; Tel: +858-784-8633
First published on 11th August 2011
Abstract
Fatty acid amide hydrolase (FAAH) is an integral membrane enzyme that degrades the endocannabinoid anandamide (AEA) and several other bioactive lipid amides. The catalytic mechanism of FAAH has been largely elucidated, and structural models of the enzyme suggest that it may recruit its hydrophobic substrates directly from the lipid bilayer of the cell. Testing this hypothesis, however, requires new tools to explore FAAH–substrate interactions in native cell membranes. Here, we have addressed this problem by creating clickable, photoreactive inhibitors that probe the microenvironment surrounding the FAAH active site. We show that these probes can be used directly in cell membranes, where distinct crosslinked adducts are observed for inhibitors that are buried within versus exposed to the external environment of the FAAH active site.
1. Introduction
Fatty acid amides are a large class of signalling lipids that regulate a diverse array of physiological processes in mammals.1,2 Representative fatty acid amides include the endogenous cannabinoid (endocannabinoid) anandamide,3 the feeding-related lipid N-oleoyl ethanolamine,4 and the sleep-inducing substance oleamide.5 Although multiple biosynthetic pathways exist for fatty acid amides,6 these lipids are degraded principally by a single enzyme—fatty acid amide hydrolase (FAAH).7–10 Genetic11 or pharmacologic12–14 inactivation of FAAH leads to elevated brain levels of many fatty acid amides, including anandamide, and produces cannabinoid receptor-dependent reductions in pain without the cognitive or locomotor defects caused by direct receptor agonists like tetrahydrocannabinol, the psychoactive component of marijuana. These findings have led to the development of several classes of FAAH inhibitors as potential therapeutic agents for treating pain and other neurological disorders.12,13,15–18FAAH is an integral membrane enzyme that appears to interact with the lipid bilayer of cells through two distinct mechanisms: 1) an N-terminal transmembrane domain, and 2) a hydrophobic patch that monotopically inserts into the membrane.19 Structural studies have revealed that the hydrophobic patch of FAAH is positioned just above the enzyme's active site, and, in certain FAAH–inhibitor structures, a continuous tunnel is observed that connects the buried catalytic triad (Ser241–Ser217–Lys142) to the membrane-interacting surface of the enzyme.20 We have hypothesized that this structural adaptation may allow FAAH to directly access and recruit its hydrophobic lipid amide substrates from cell membranes into the enzyme's active site.
The aforementioned hypothesis has, however, remained untested due to a lack of tools and methods for interrogating the microenvironment surrounding the FAAH active site. For such an approach to succeed, it would ideally enable the characterization of FAAH in native membrane preparations. Here, we have addressed this problem by creating clickable, photoreactive inhibitors of FAAH. We show that these probes can be used directly in cell membranes, and find that inhibitors of varying length produce distinct crosslinked adducts depending on whether they are buried within or exposed to the external environment surrounding the FAAH active site.
2. Experimental procedures
2.1 Generation of FAAH and TAP(CBP/FLAG)-FAAH constructs
Mouse FAAH in the pcDNA vector was generated as described previously.7 The coding sequence for the calmodulin binding peptide and FLAG tags were synthesized (Integrated DNA Technologies) and cloned at the N-terminus of FAAH in the pcDNA3 vector giving TAP(CBP/FLAG)-FAAH.2.2 Recombinant expression of FAAH and TAP(CBP/FLAG)-FAAH proteins in COS-7 cells and cell membrane preparation
Briefly, COS-7 cells were grown to ∼70% confluencey in 10 cm dishes in complete medium (DMEM with L-glutamine, nonessential amino acids, sodium pyruvate, and FCS) at 37 °C and 5% CO2. The cells were transiently transfected using pcDNA3 encoding mouse FAAH or TAP(CBP/FLAG)-FAAH using the FUGENE 6 (Roche Applied Science) transfection reagent according to the manufacturer's protocols. After two days, cells were washed twice with phosphate-buffered saline (PBS; pH 7.4), collected by scraping, re-suspended in 1.0 mL PBS, and pelleted by centrifugation at 5000 rpm for 5 min at 4 °C. The resulting supernatant was discarded and the cells re-suspended in PBS and lysed by sonication. The lysed cells were centrifuged at 100,000 × g for 45 min at 4 °C, the supernatant was discarded and the pellet was re-suspended in PBS by sonication. Protein concentrations were measured by using the Bio-Rad DC Protein Assay Kit, and aliquots were stored at −80 °C until use.2.3 In vitro inhibition potency studies
Inhibitor analysis was carried out as described previously.21,22 Briefly, cell membranes (1 mg mL−1 in PBS, pH 8, 50 μl) were pre-incubated with varying concentrations of probe (1 μl of 50× stock in DMSO added to provide 0.002–20 μM final concentration) for 10 min at RT. 14C-Oleamide (1.25 μl, 4 mM stock in DMSO, 100 μM final concentration) was added, incubated for 5 min, and the reaction quenched with 400 μl of 0.5 M HCl. The solution was then extracted with 600 μl of ethyl acetate. The organic layer was removed and dried under a stream of gaseous N2, solubilized in 10 μl of ethyl acetate, and separated by TLC (60% ethyl acetate in hexanes). The radioactive compounds were quantified using a Cyclone Phosphorimager (PerkinElmer Life Sciences).2.4 Competitive ABPP and click-chemistry studies
Competitive ABPP and click-chemistry reactions were performed as previously described.22 Briefly, cell membranes (100 μg of protein in 50 μl of PBS) incubated with 2 μM probe (1 μl of 100 μM DMSO stock) for 1 h at RT, followed by 5 μM rhodamine-tagged fluorophosphonate23,24 (FP–Rh; 1 μl of 250 μM stock in DMSO) for 1 h at RT. For click-chemistry reaction, membranes were first treated with 2 μM probe followed by click chemistry reaction with 25 μM rhodamine-azide (RhN3, 1 μl of 1.25 mM DMSO stock). Reactions were quenched with an equal volume (50 μl) of 2 × SDS loading buffer, separated by SDS-PAGE, and visualized by in-gel fluorescence scanning with a Hitachi FMBio IIe flatbed fluorescence scanner (MiraiBio).2.5 Crosslinking studies
Cell membranes (400 μg of protein in 200 μl of PBS) were incubated with 2 μM probe (4 μl of a 100 μM DMSO stock) for 1 h at RT with or without PF-3845 (2 μM) pre-treatment (1 h at RT). Reaction mixtures were treated either with or without UV irradiation (365 nm) for 1 h, followed by click chemistry reaction with 25 μM RhN3 as described above. Proteins were precipitated as follows: four volume of methanol and one volume of chloroform were added, samples were vortex-mixed and three volume of water was added. After vortex-mixing and centrifugation at 14,000 rpm (∼9000 × g) for 3 min, the top and bottom layers were removed leaving behind the protein interface. The protein pellet was re-suspended in 500 μl of methanol by sonication and pelleted by centrifugation at 14,000 rpm for 3 min. Methanol was removed, 50 μl of 25 mM ammonium bicarbonate/6M urea (aq.) was added and samples were heated at 65 °C for 5 min. After denaturation, proteins were treated with 10 mM DTT (0.5 μl of 1 M DTT in water) for 15 min at 65 °C to reduce disulfides. Cysteines were alkylated by incubating samples with 30 mM iodoacetamide (3 μl of 0.5 M iodoacetamide in water) for 30 min in dark at RT. The urea concentration of samples was diluted to 2 M with 25 mM ammonium bicarbonate and samples were treated with or without trypsin (16 μg ml−1) or endoproteinase Lys-C (3 μg ml−1) for 3 h at 37 °C. After digestion, samples were diluted with 2 × Tricine SDS sample buffer, run on Novex® 10–20% Tricine Gel 1.0 mm (Invitrogen) and visualized by in-gel fluorescence scanning with a Hitachi FMBio IIe flatbed fluorescence scanner (MiraiBio).2.6 Mass spectrometry studies
3. Results and discussion
3.1 Chemical probe design and synthesis
Building on the success of activity-based probes that contain electrophilic and/or photoreactive groups for enzyme labeling,29 we sought to incorporate four elements into our active site probes for FAAH: a carbamate electrophile (1) for covalent labeling of the enzyme's catalytic serine nucleophile22 connected through a variable-length linker (2) to a benzophenone photocrosslinker30,31 (3) and a clickable alkyne group30,31 (4) (Fig. 1). Once covalently bonded to the FAAH serine nucleophile, probe-enzyme adducts would be exposed to UV light to induce crosslinking to proximal molecular species. Click chemistry to suitable reporter tags (fluorophore, biotin) could then be used to visualize, enrich, and characterize the crosslinked products.32,33 By comparing the structures of these crosslinked adducts for probes of varying length, the environment surrounding the FAAH active site could be systematically explored. Toward this end, we synthesized four clickable, photoreactive inhibitors (Fig. 1) with increasing linker lengths between the carbamate and benzophenone groups (see Supporting Information for synthesis†). Briefly, 4,4′-diaminobenzophenone was reacted with a Boc-protected amino fatty acid (1a–d, Scheme S1†) and the resulting compound was coupled with 5-hexynoic acid using DCC/DMAP (HOBt). After Boc-deprotection using TFA, the amino group was reacted with phenyl chloroformate to give the final compound (5a–d).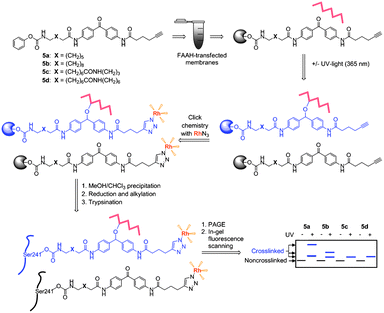 | ||
| Fig. 1 Schematic for crosslinking studies with probes 5a–d. | ||
3.2 FAAH inhibition
Compounds 5a–d were all found to inhibit FAAH with good potency as determined by a radiolabeled substrate assay,34 showing IC50 values between 200–750 nM (Fig. 2a). For use in complex proteomes, the probes also needed to show selectivity for FAAH relative to other serine hydrolases, which have the potential to react with carbamates. Competitive activity-based protein profiling (ABPP)35 was used to assess the specificity of compounds 5a–d for FAAH. Membrane proteomes from COS-7 cells transfected with FAAH were treated with 5a–d and then reacted with fluorophosphonate-rhodamine (FP–Rh),24 which serves as a general ABPP probe for serine hydrolases.36 Compounds 5a–d completely and selectively blocked FP–Rh labeling of FAAH at 2 μM when compared to mock and DMSO treated membranes (Fig. 2b, left). To further confirm the selectivity of 5a–d for FAAH, 5a-d-treated cell membranes were reacted with rhodamine-azide (RhN3) under click chemistry conditions.32,33FAAH was observed to be the primary target of 5a–d in COS-7 cell membranes (Fig. 2b, right).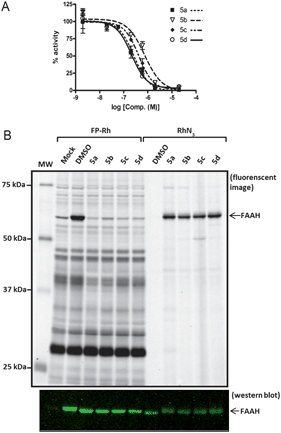 | ||
| Fig. 2 Potency and selectivity of probes 5a–d as inhibitors of FAAH. a) Concentration-dependent inhibitor curves for probes 5a–d. Varying concentrations of 5a–d were pre-incubated with cell membranes for 10 min. 14C-Oleamide was added, reaction was quenched in 0.5 M HCl at 5 min, and the solution was extracted with ethyl acetate. The organic layer was removed, dried, re-solubilized, and separated by TLC. The radioactive compounds were quantified using a Cyclone Phosphorimager. The data represent the mean ± SEM of an experiment conducted in duplicate. IC50 values were calculated using nonlinear regression sigmoidal dose-response-variable slope. b) Competitive ABPP with FP–Rh and click-chemistry with RhN3. COS-7 cell membranes were treated with 5a–d (2 μM), followed by FP–Rh (left lanes) or click chemistry reaction with RhN3 (right lanes). Reactions were quenched with 2 × SDS loading buffer, separated by SDS-PAGE, and visualized by in-gel fluorescence scanning (top; image shown in grey-scale) or Western blotting (bottom) with anti-FAAH rabbit polyclonal antibodies. Western blot confirms equal loading of FAAH for both experiments. | ||
3.3 Gel-based crosslinking studies
Having confirmed that 5a–d were potent and selective FAAH inhibitors, we next investigated their photocrosslinking profiles in COS-7 cell membranes expressing FAAH (Fig. 3). In this assay, membrane proteomes from FAAH-transfected cells were treated with 5a–d and exposed to long-wave UV light to induce crosslinking. Probes were conjugated to RhN3 under click chemistry reaction conditions, and proteomes were either directly analyzed by SDS-PAGE and in-gel fluorescence scanning or first precipitated with methanol/chloroform to remove lipids and excess click reagents and then treated with trypsin (Fig. 3, lanes 5–8, all panels) or endoproteinase Lys–C (Fig. 3, lanes 9–12, all panels) prior to SDS-PAGE analysis (using 10–20% Tricine gels). As observed previously (Fig. 2b), a 65 kDa band representing FAAH was observed in non-digested samples (Fig. 3, lanes 3–4, all panels). In the photocrosslinked samples (Fig. 3, lane 4, all panels), FAAH appeared to migrate at a slightly higher molecular weight with an intensity largely unaffected by crosslinking, suggesting that reaction occurred in an intramolecular fashion (to FAAH itself) or in an intermolecular manner to a small molecule (< ∼2 kDa). We also observed a probe-reactive 75 kDa protein in undigested, crosslinked samples, but this band was not inhibited by PF-3845 (see, for example, Fig. 3 lanes 2 and 4) so we believe that it reflects a non-specific target of the probes.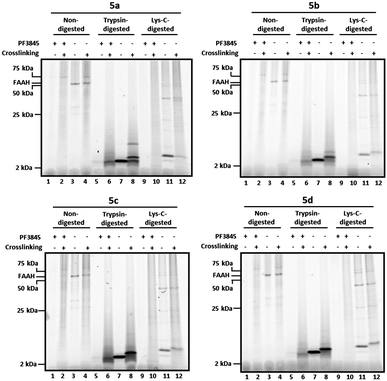 | ||
| Fig. 3 Tricine gels of crosslinked and noncrosslinked samples treated with or without trypsin or Lys–C protease. Briefly, FAAH-transfected COS-7 cell membranes were incubated with 2 μM 5a–d. As a control, membranes pre-treated with the FAAH inhibitor PF-3845 (2 μM) are shown for comparison (lanes 1–2, 5–6, and 9–10, all panels). Reaction mixtures were treated with or without (alternating lanes) UV irradiation (365 nm) and subjected to click chemistry with RhN3. For digested samples, proteins were precipitated with methanol/chloroform prior to reduction, alkylation, and digestion with either trypsin (lanes 5–8, all panels) or Lys–C (lanes 9–12, all panels). Samples were separated on 10–20% Tricine gels and visualized by in-gel fluorescence scanning (fluorescent images shown in grey scale). See Fig. 4 for enlarged views of trypsin- and Lys-C-digested bands in the 2–25 kDa range. | ||
In noncrosslinked trypsin-digested samples (Fig. 3, lane 7, all panels; see also Fig. 4a, lane 3, all top panels), a single probe-labeled band was observed at ∼3.7 kDa, which corresponds to the predicted mass of FAAH's active site tryptic peptide modified with 5a–d and the Rh tag (theoretical masses: 3.66–3.80 kDa for 5a–d). In the photocrosslinked, trypsin-digested samples, the active-site peptide band migrated as one or more higher molecular weight bands (Fig. 3, lane 8, all panels; see also Fig. 4a, lane 4, all top panels). For 5b–d, the uncrosslinked ∼3.7 kDa peptide appears to be largely absent in the photocrosslinked samples (Fig. 3, lane 8, panels 5b–d; see also Fig. 4a, lane 4, top panels 5b–d); in contrast, for 5a, this uncrosslinked peptide was still observed in photocrosslinked samples (Fig. 3, lane 8, panel 5a; see also Fig. 4a, lane 4, top panel 5a), indicating incomplete crosslinking for the shortest probe. A much higher molecular weight crosslinked product was also observed specifically for 5a, but not the other probes (see Fig. 4b for direction comparison). This may represent an internal crosslinking event within the FAAH protein that links two tryptic peptides together, as has been observed in previous photocrosslinking studies aimed at mapping the FAAH active site.37 Consistent with this premise, modeling suggests that probe 5a is the only probe within the 5a–5d series that would place the benzophenone group within the active site of FAAH (Fig. 5).
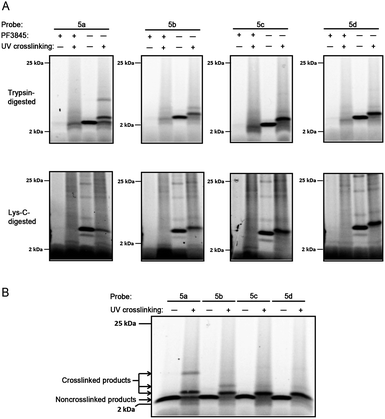 | ||
| Fig. 4 Tricine gels of crosslinked and noncrosslinked samples. a) Enlarged images of trypsin-digested (top) and Lys-C-digested (bottom) samples from Fig. 3, 2–25 kDa range. Briefly, FAAH-transfected COS-7 cell membranes were incubated with 2 μM probe 5a–d. As a control, membranes pre-treated with the FAAH inhibitor PF-3845 (2 μM) are shown for comparison (lanes 1–2, all panels). Reaction mixtures were treated with or without (alternating lanes) UV irradiation (365 nm) and subjected to click chemistry with RhN3. For digested samples, proteins were precipitated with methanol/chloroform prior to reduction, alkylation, and digestion with either trypsin (top) or Lys–C (bottom). Samples were separated on 10–20% Tricine gels and visualized by in-gel fluorescence scanning (fluorescent images shown in grey scale). b)Tricine gel of noncrosslinked and crosslinked trypsin-digested samples for 5a–d run side-by-side. Sample preparation and analysis same as for a, above. | ||
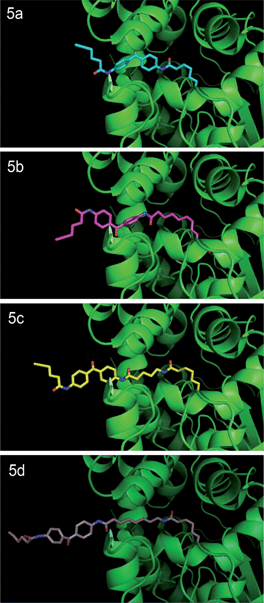 | ||
| Fig. 5 Probes 5a–d docked into the three-dimensional structure of rat FAAH. Crystal structure of humanized-rat FAAH conjugated with ligand OL135 (PDB: 2WJ2) was used as starting template to manually build FAAH-probe docking models using PyMOL. Initial three-dimensional structures of Probes 5a–d were generated by ChemDraw 12.0. The first alkyl linker connecting the carbamate group in Probes 5a–d was structurally aligned with the alkyl linker of the OL1 ligand in the crystal structure, which enables the head (carbamate) of each probe to be docked in the ligand binding channel of native rat FAAH. Dihedral angles of carbon–carbon bonds within the first and second alkyl linker regions were then adjusted in preferred rotameric states to release steric clash between the tail (benzophenone) of the probe and the protein. | ||
Similar trends were observed for Lys–C digested samples. In noncrosslinked samples (Fig. 3, lane 11, all panels; see also Fig. 4a, lane 3, bottom panels), the dominant band was observed to migrate at ∼5 kDa, which corresponds to the predicted mass of the 5a-d-modified/Rh-conjugated FAAH active site peptide (theoretical masses: 5.01–5.15 kDa for 5a–d). In the crosslinked, Lys-C-digested samples for 5b–d (Fig. 3, lane 12, panels 5b–5d; see also Fig. 4a, lane 4, bottom panels), the dominant band is shifted to higher MW with concurrent disappearance of the uncrosslinked peptide band. The shortest probe, 5a, was again observed to behave differently than its counterparts with longer linkers; a light band corresponding to the uncrosslinked peptide was the only significant band in the UV-treated sample (Fig. 3, lane 12, panel 5a versus 5b-5d; see also Fig. 4a, lane 4, bottom panel 5a versus 5b-5d). Importantly, none of the putative probe-labeled FAAH protein/peptide bands were present in samples pre-treated with the selective FAAH inhibitor PF-384513 (Fig. 3, lanes 1–2, 5–6, 9–10, all panels; see also Fig. 4a, lanes 1–2, all panels), confirming that they originate from active site-labeling of FAAH rather than cross reactivity with another cellular protein.
Taken together, these data indicate that the longer active-site probes 5b–d produce a similar photocrosslinking profile that differs substantially from the profile generated with the shorter active-site probe 5a. These data are consistent with modelling studies indicating that probes 5b–d project the benzophenone crosslinker out of the FAAH active site, possibly into the cell membrane, while the benzophenone group of shortest probe 5a is predicted to remain within the FAAH active site (note that probe 5a is ∼7 angstroms shorter than probe 5b and this is about the same distance between the benzophenone carbonyl of 5a and the surface of the enzyme active site that is predicted to interface with the membrane). That a similar mass shift is observed for the products of 5b–d when FAAH is digested with either tryspin or Lys–C suggests that the crosslinked biomolecule may be non-proteinaceous in nature.
3.4 Mass spectrometry-based crosslinking studies
We next used liquid chromatography-tandem mass spectrometry (LC-MS/MS) to characterize the probe-labeled active site peptides before and after crosslinking. COS-7 cell membranes expressing a FLAG epitope-tagged FAAH were incubated with probes 5a–d and then treated with or without UV irradiation to effect crosslinking. FAAH proteins were enriched by anti-FLAG antibody beads and trypsin digested on the solid support. The released peptides were then analysed by LC-MS/MS. Quantification of the integrated peak of the probe-labeled active site FAAH peptide revealed a dramatic decrease in its abundance after photocrosslinking as shown in Fig. 6 for probe 5a (see Supplemental Figure S1 for complete analysis of all probes†). Due to the length and hydrophobicity of the probe-labeled peptides, they were observed to elute at the very end of the chromatographic gradient (95–100% Buffer B), even with LC conducted at 60 °C. The elution profile was not altered by varying the gradient or using larger pore size reversed-phase resins (data not shown). Because any crosslinked products would be larger and likely even more hydrophobic than the noncrosslinked species we were not surprised that these crosslinked adducts were unobservable using our sample analysis conditions; indeed, a thorough search for modified peaks by comparative LC analysis and MS extraction of theoretical products failed to identify crosslinked species (data not shown). Future analysis of crosslinked products will involve use of alternate digestion strategies to reduce the length (and thus retention time) of the labeled active site peptide and resultant crosslinked products, as well as other possible modifications to the chromatography procedure to facilitate characterization of very large and hydrophobic modified peptides.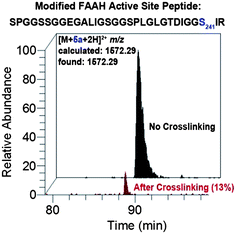 | ||
| Fig. 6 LC-MS analysis showing loss of probe-modified FAAH active site peptide following crosslinking for probe 5a. Extracted ion chromatograms correspond to the probe-labeled active site FAAH peptide before (black) and after (red) photocrosslinking. Relative peak area after crosslinking is indicated in parenthesis. The probe-modified residue is catalytic Ser241. See Supplemental Figure S1 for LC-MS analysis of all probes. Note: peak identities and site-of-labeling (Ser241) were confirmed by SEQUEST26 analysis of tandem mass spectra as summarized in Supplemental Table S1. | ||
Conclusions
Understanding how enzymes interact with their physiologic substrates in native cellular environments is an important problem. This is especially pertinent for integral membrane enzymes, which often perform chemistry on hydrophobic substrates (e.g., lipids, other transmembrane proteins) that are themselves embedded within the lipid bilayer. It seems likely that, in these instances, enzymes would have evolved an intricate mechanism for interacting with the cell membrane to access their substrates and release products. Nonetheless, there are few methods to probe enzyme–membrane interactions in complex biological systems. Here, we have presented a chemoproteomic strategy that addresses this problem for the integral membrane enzyme FAAH. We generated a set of covalent FAAH inhibitors that also possess photocrosslinking and clickable groups and used these reagents to probe the microenvironment outside of the FAAH active site. By varying the length of the probes, we generated data that support selective intermolecular crosslinking to a small-molecular weight product(s) for the longer probes that are modelled to protrude from the FAAH active site. While we do not yet know the identity of this crosslinked product, it may represent a lipid constituent of the membrane. Future studies will attempt to address this question, but will likely require extensive optimization of protease-digestion and LC-MS protocols to generate a crosslinked product that can be detected and characterized.The method described herein should be applicable to many other enzymes, especially those for which covalent inhibitors are available. Indeed, Overkleeft and colleagues recently reported an investigation of proteasomal subunit interactions using photoreactive, clickable inhibitors.38 Our studies thus extend this active-site crosslinking approach to a membrane enzyme and show that it is amenable to complex proteomic environments from transfected cells. Conceivably, the method could also be applied to membrane preparations from tissues that naturally express FAAH (e.g., brain, liver), since the active site probes can be fully functionalized to enable labeling, photocrosslinking, and coordinated detection and affinity enrichment (by clicking to ‘trifunctional’ biotin/fluorphore azide tags32) of enzymes. The continued advancement of chemoproteomic methods for characterizing active site interactions should greatly enrich our understanding of how enzymes function in native biological environments.
Acknowledgements
This work was supported by National Institutes of Health Grant DA017259 (B.F.C), Academy of Finland Grant 130650, University of Kuopio (S.M.S), and the Skaggs Institute for Chemical Biology.Notes and references
- V. Di Marzo, T. Bisogno and L. De Petrocellis, Chem. Biol., 2007, 14, 741–756 CrossRef CAS.
- C. J. Fowler, S. Holt, O. Nilsson, K. O. Jonsson, G. Tiger and S. O. Jacobsson, Pharmacol., Biochem. Behav., 2005, 81, 248–262 CrossRef CAS.
- W. A. Devane, L. Hanus, A. Breuer, R. G. Pertwee, L. A. Stevenson, G. Griffin, D. Gibson, A. Mandelbaum, A. Etinger and R. Mechoulam, Science, 1992, 258, 1946–1949 CAS.
- F. Rodriguez de Fonseca, M. Navarro, R. Gomez, L. Escuredo, F. Nava, J. Fu, E. Murillo-Rodriguez, A. Giuffrida, J. LoVerme, S. Gaetani, S. Kathuria, C. Gall and D. Piomelli, Nature, 2001, 414, 209–212 CrossRef CAS.
- B. F. Cravatt, O. Prospero-Garcia, G. Siuzdak, N. B. Gilula, S. J. Henriksen, D. L. Boger and R. A. Lerner, Science, 1995, 268, 1506–1509 CAS.
- G. M. Simon and B. F. Cravatt, Mol. BioSyst., 2010, 6, 1411–1418 RSC.
- B. F. Cravatt, D. K. Giang, S. P. Mayfield, D. L. Boger, R. A. Lerner and N. B. Gilula, Nature, 1996, 384, 83–87 CrossRef CAS.
- K. Ahn, M. K. McKinney and B. F. Cravatt, Chem. Rev., 2008, 108, 1687–1707 CrossRef CAS.
- D. G. Deutsch, N. Ueda and S. Yamamoto, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2002, 66, 201–210 CrossRef CAS.
- C. J. Fowler, K. O. Jonsson and G. Tiger, Biochem. Pharmacol., 2001, 62, 517–526 CrossRef CAS.
- B. F. Cravatt, K. Demarest, M. P. Patricelli, M. H. Bracey, D. K. Giang, B. R. Martin and A. H. Lichtman, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 9371–9376 CrossRef CAS.
- S. Kathuria, S. Gaetani, D. Fegley, F. Valino, A. Duranti, A. Tontini, M. Mor, G. Tarzia, G. La Rana, A. Calignano, A. Giustino, M. Tattoli, M. Palmery, V. Cuomo and D. Piomelli, Nat. Med., 2003, 9, 76–81 CrossRef CAS.
- K. Ahn, D. S. Johnson, M. Mileni, D. Beidler, J. Z. Long, M. K. McKinney, E. Weerapana, N. Sadagopan, M. Liimatta, S. E. Smith, S. Lazerwith, C. Stiff, S. Kamtekar, K. Bhattacharya, Y. Zhang, S. Swaney, K. Van Becelaere, R. C. Stevens and B. F. Cravatt, Chem. Biol., 2009, 16, 411–420 CrossRef CAS.
- S. Holt, F. Comelli, B. Costa and C. J. Fowler, Br. J. Pharmacol., 2005, 146, 467–476 CrossRef CAS.
- A. H. Lichtman, D. Leung, C. Shelton, A. Saghatelian, C. Hardouin, D. Boger and B. F. Cravatt, J. Pharmacol. Exp. Ther., 2004, 311, 441–448 CrossRef CAS.
- D. L. Boger, H. Sato, A. E. Lerner, M. P. Hedrick, R. A. Fecik, H. Miyauchi, G. D. Wilkie, B. J. Austin, M. P. Patricelli and B. F. Cravatt, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 5044–5049 CrossRef CAS.
- K. Ahn, D. S. Johnson, L. R. Fitzgerald, M. Liimatta, A. Arendse, T. Stevenson, E. T. Lund, R. A. Nugent, T. K. Nomanbhoy, J. P. Alexander and B. F. Cravatt, Biochemistry, 2007, 46, 13019–13030 CrossRef CAS.
- B. Koutek, G. D. Prestwich, A. C. Howlett, S. A. Chin, D. Salehani, N. Akhavan and D. G. Deutsch, J. Biol. Chem., 1994, 269, 22937–22940 CAS.
- M. H. Bracey, M. A. Hanson, K. R. Masuda, R. C. Stevens and B. F. Cravatt, Science, 2002, 298, 1793–1796 CrossRef CAS.
- M. Mileni, D. S. Johnson, Z. Wang, D. S. Everdeen, M. Liimatta, B. Pabst, K. Bhattacharya, R. A. Nugent, S. Kamtekar, B. F. Cravatt, K. Ahn and R. C. Stevens, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12820–12824 CrossRef CAS.
- J. P. Alexander and B. F. Cravatt, J. Am. Chem. Soc., 2006, 128, 9699–9704 CrossRef CAS.
- J. P. Alexander and B. F. Cravatt, Chem. Biol., 2005, 12, 1179–1187 CrossRef CAS.
- N. Jessani, Y. Liu, M. Humphrey and B. F. Cravatt, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 10335–10340 CrossRef CAS.
- M. P. Patricelli, D. K. Giang, L. M. Stamp and J. J. Burbaum, Proteomics, 2001, 1, 1067–1071 CrossRef CAS.
- A. E. Speers, A. R. Blackler and C. C. Wu, Anal. Chem., 2007, 79, 4613–4620 CrossRef CAS.
- J. Eng, A. L. McCormack and J. R. Yates, J. Am. Soc. Mass Spectrom., 1994, 5, 976–989 CrossRef CAS.
- A. Keller, A. I. Nesvizhskii, E. Kolker and R. Aebersold, Anal. Chem., 2002, 74, 5383–5392 CrossRef CAS.
- D. L. Tabb, W. H. McDonald and J. R. Yates, J. Proteome Res., 2002, 1, 21–26 CrossRef CAS.
- B. F. Cravatt, A. T. Wright and J. W. Kozarich, Annu. Rev. Biochem., 2008, 77, 383–414 CrossRef CAS.
- S. A. Sieber, S. Niessen, H. S. Hoover and B. F. Cravatt, Nat. Chem. Biol., 2006, 2, 274–281 CrossRef CAS.
- C. M. Salisbury and B. F. Cravatt, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1171–1176 CrossRef CAS.
- A. E. Speers and B. F. Cravatt, Chem. Biol., 2004, 11, 535–546 CrossRef CAS.
- A. E. Speers, G. C. Adam and B. F. Cravatt, J. Am. Chem. Soc., 2003, 125, 4686–4687 CrossRef CAS.
- M. K. McKinney and B. F. Cravatt, J. Biol. Chem., 2003, 278, 37393–37399 CrossRef CAS.
- D. Leung, C. Hardouin, D. L. Boger and B. F. Cravatt, Nat. Biotechnol., 2003, 21, 687–691 CrossRef CAS.
- D. A. Bachovchin, T. Ji, W. Li, G. M. Simon, J. L. Blankman, A. Adibekian, H. Hoover, S. Niessen and B. F. Cravatt, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20941–20946 CrossRef CAS.
- M. P. Patricelli and B. F. Cravatt, Biochemistry, 2001, 40, 6107–6115 CrossRef CAS.
- P. P. Geurink, B. I. Florea, G. A. Van der Marel, B. M. Kessler and H. S. Overkleeft, Chem. Commun., 2010, 46, 9052–9054 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthesis and characterization of probes 5a–d, LC-MS analysis (extracted chromatograms) of crosslinked and noncrosslinked peptides, and LC-MS/MS analysis (SEQUEST) of crosslinked and noncrosslinked peptides. See DOI: 10.1039/c1sc00336d |
| This journal is © The Royal Society of Chemistry 2012 |